서론
사이질폐질환(interstitial lung diseases, ILD)은 폐 사이질의 증식과 함께 다양한 염증세포들의 침윤과 함께 때로는 섬유화가 동반되어 비정상적인 콜라젠 침착을 나타내는 질환들을 말한다[1]. ILD는 그 원인에 따라 직업환경성, 의인성, 결체조직질환, 특발성의 네 가지로 분류할 수 있다. 하지만 이런 원인에 따른 분류로는 질환의 임상 양상을 제대로 설명할 수 없다는 단점이 있어, 2013년 개정된 American Thoracic Society/European Respiratory Society guidelines에서는 질환의 개선 가능성 및 진행성 여부에 따라 사이질폐질환을 나누어 분류하기도 하였다[2]. 그 중에서도 최근에는 진행성 표현형을 나타내는 만성 섬유성 사이질폐질환 (chronic fibrosing ILD with a progressive phenotype, PF-ILD)의 진행을 막기 위한 여러 치료법에 대한 연구가 활발하게 진행되고 있다. 이에 이 논문에서는 PF-ILD의 개념과 치료법에 대하여 소개하고 앞으로 나아가야할 방향에 대해서도 알아보고자 한다.
PF-ILD의 개념
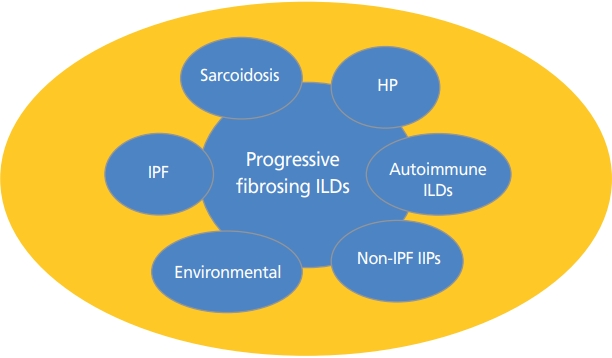
일반적으로 ILD는 그 원인에 따라 분류되어 왔으며, 그 중에서 특발성폐섬유증(idiopathic pulmonary fibrosis, IPF)이 가장 흔한 것으로 보고되어 왔다. IPF는 폐기능의 감소와 짧은 평균수명을 특징으로 하는 대표적인 진행성 섬유성 ILD라 할 수 있다[2]. 그러나 IPF 외에도 비특이사이질 폐렴, 분류할 수 없는 특발성사이질폐렴, 자가면역관련 ILD, 사르코이드증(sarcoidosis), 과민성폐렴, 직업환경성 ILD 등으로 구분된 다른 ILD 또한 IPF와 유사한 진행성의 섬유성 ILD의 임상양상을 보이는 경우가 많다는 것이 보고되었다(Figure 1) [3]. 폐 섬유화의 진행은 호흡기적 증상과 폐기능, 삶의 질의 악화를 유발할 뿐만 아니라 사망률 또한 증가시킬 수 있으므로 이에 대한 조기 발견 및 적절한 관리가 필요하다[4,5]. 그러므로 원인에 따라 ILD를 분류하는 기존의 분류법 이외에도, 치료에 반응하지 않고 진행하는 섬유화를 보이는 여러 ILD를 하나의 임상적인 질병군으로 분류하여야 할 필요성이 대두되고 있다. 이에 따라 기존의 치료에도 불구하고 영상학적으로 진행하는 폐섬유화, 시간이 지남에 따라 악화되는 호흡기 증상, 폐기능의 점진적인 악화, 높은 사망률 등의 공통적인 임상 표현형을 보이는 ILD를 PF-ILD라 통칭하게 되었으며, 최근 이에 대한 연구가 활발해지고 있다(Table 1) [6]. 다음 장에서는 우선 PF-ILD의 특징적인 정의에 대해 알아보고자 한다.
PF-ILD의 정의
실제 임상에서 ILD의 진행은 환자의 증상, 폐기능 및 흉부 이미지 검사를 통하여 확인할 수 있다. 그 중에서도 폐기능은 ILD의 진행을 판단하는데 있어 필수적인 요소로서, 강제폐활량(forced vital capacity, FVC) 및 일산화탄소확산능(diffusing capacity of the lung for carbon monoxide, DLCO)의 감소를 관찰함으로써 ILD의 진행을 확인할 수 있다. 현재까지 PF-ILD에서 폐기능 검사 결과 중 어떤 지표를 섬유화의 진행으로 판단할 지에 대한 학계의 의견 일치는 없는 상태이나, 여러 연구에서는 일반적으로 FVC 혹은 DLCO가 10% 이상 감소하는 것을 섬유화의 진행으로 정의하고 있다[7-12]. 하지만 FVC 와 DLCO 등의 폐기능 검사 결과는 그 변화를 예측하기 힘들 뿐만 아니라, 변동성이 심하여 폐기능 검사만으로 PF-ILD를 진단하기에는 한계가 있다[13].
ILD의 진단에 있어서 흉부 컴퓨터단층촬영(computed tomography, CT)은 필수적인 도구 중 하나이다. 특히 고해상도 CT (high-resolution CT, HRCT)는 ILD의 진단에 있어 일반적인 CT보다 더욱 민감한 것으로 알려져 있으며 여러 가이드라인에서도 ILD의 진단을 위해 HRCT의 촬영을 권고하고 있다[14]. 최초의 흉부 HRCT에서 보통사이질폐렴(usual interstitial pneumonia, UIP) 소견이 보이는 경우 폐 섬유화가 진행할 가능성이 높다고 볼 수 있으나, 흉부 HRCT 소견은 ILD의 원인에 따라 다양한 양상으로 나타나므로 UIP 소견이 없다고 해서 PF-ILD를 배제할 수는 없다[15]. 그러므로 PF-ILD를 진단하기 위해서는 연속적으로 HRCT를 촬영하여 벌집모양(honeycombing)과 망상음영(reticular opacity) 등의 섬유화를 시사하는 소견이 시간이 지남에 따라 증가하는 지에 대한 여부를 판단하여야 한다.
섬유화 진행의 위험인자
PF-ILD에 관한 여러 후향적 연구를 통해 남성, 고령, 진단 당시 낮은 FVC와 DLCO 등이 PF-ILD의 진행 및 불량한 예후와 연관이 있음이 밝혀졌다[11,20-22]. 특히 영상학적 또는 조직학적 UIP 소견의 존재가 높은 사망률과 연관이 있는 것으로 알려져 있는데, ILD를 유발하는 원인과 상관없이 UIP의 존재 자체가 ILD의 진행 및 사망률의 증가와 연관이 있으므로 UIP가 있는 환자의 경우에는 문진, 폐기능 검사, 흉부 영상 등을 통해 섬유화의 진행에 대해 면밀하게 관찰해야 한다[22-24].
위식도 역류성 질환(gastroesophageal reflux disease, GERD) 또한 반복적인 미세흡인을 통해 ILD에서 섬유화의 진행을 일으킬 수 있는 것으로 알려져 있다[25]. 특히 전신경화증 환자에서는 GERD가 폐섬유화 발생의 중요한 원인 중 하나로 고려되고 있으며[26,27], IPF 환자에서는 GERD의 위험인자로 알려져 있는 틈새탈장(hiatal hernia)이 폐기능의 감소 및 사망률의 증가와 연관되어 있는 것으로 보고되고 있다[28,29]. 그러나 현재까지 GERD가 전신경화증관련 ILD (systemic sclerosis-related ILD, SSc-ILD)나 IPF 이외의 다른 PF-ILD를 악화시킬 수 있다는 명확한 근거는 없기에, 추후에 GERD와 다른 PF-ILD의 진행에 대한 전향적 연구결과를 지켜보아야 할 것이다.
유전학적으로는 끝분절효소(telomerase)의 기능부전이 폐섬유화의 진행에 중요한 역할을 할 수 있다[30,31]. 끝분절 효소는 DNA의 증식 중에 발생하는 단축을 상쇄시키는 역할을 하는데, 이 효소의 기능에 이상이 발생하면 세포가 증식하여 한계 길이에 도달하면 끝분절(telomere)이 짧아짐에 따라 DNA 손상반응을 촉발하여 세포노화와 세포자멸사를 유발하고 줄기세포의 재생 잠재력을 감소시키게 된다[32]. 이로 인해 폐포 상피세포의 노화가 촉진되고 재상피화가 원활하지 못하게 되어 폐 섬유화를 진행시킬 수 있다. 끝분절의 기능저하와 연관된 가장 흔한 ILD는 IPF로, 가족성 IPF 환자의 약 1/3은 끝분절이 짧아져 있거나 끝분절과 관련된 유전자 돌연변이를 가지고 있는 것으로 보고되고 있다[33]. IPF가 아닌 ILD에서도 끝분절의 이상이 있는 환자는 평균적으로 연간 300 mL 이상의 FVC 감소와 평균 3년 정도의 짧은 생존기간을 보여주었으며[34], 만성 과민성폐렴 환자의 경우 끝분절 또는 끝분절효소의 기능부전이 있는 환자는 그렇지 않은 환자에 비해 불량한 예후와 더 심한 폐 섬유화가 나타나는 경향을 보여주었다[31,35]. 끝분절의 이상을 확인하기 위해서는 말초 혈액에서 백혈구의 끝분절 길이를 측정하거나 끝분절효소와 관련된 유전자 돌연변이를 확인할 수 있으나, 현재 임상에서 이러한 검사들은 현실적으로 사용이 어려운 실정이다. 그러나 이러한 검사를 통하여 끝분절의 이상을 미리 알게 된다면, 조기에 항섬유화제의 사용을 고려하고 이를 통해 환자의 예후를 개선할 수 있을 가능성이 있으므로 끝분절 이상의 조기발견에 대한 보다 활발한 연구가 필요할 것으로 생각된다.
PF-ILD의 치료
PF-ILD의 치료는 크게 비약물적 치료와 약물적 치료로 나눌 수 있다. 비약물적 치료는 다른 ILD에 대한 치료와 크게 다르지 않다. 흡연을 지속하는 것이 섬유화의 진행에 미치는 영향에 대해서는 아직 논란의 여지가 있으나, 흡연은 직간접적으로 환자의 폐기능을 악화시키고 폐암의 발병률을 높일 수 있으므로 흡연 중인 모든 ILD 환자에게 금연을 권고해야 한다[36]. 직업성/약물 유발성 ILD 및 과민성 폐렴의 경우에는 ILD를 유발할 수 있는 인자에 대한 노출을 중단하는 것이 중요하다. 중증의 ILD 환자의 경우 근육의 감소를 동반한 신체활동의 감소가 흔하게 나타나므로 호흡재활을 통하여 환자의 증상과 운동능력을 개선시킬 수 있다[37,38]. 또한 폐이식의 적응증에 해당하는 환자에 대해서는 적극적으로 조기에 폐이식에 대한 평가를 시행하는 것이 삶의 질 개선 및 생존율 향상에 기여할 수 있을 것으로 생각된다[39]. PF-ILD의 약물적 치료는 크게 항섬유화제와 면역억제제로 나누어 볼 수 있다. 다음 장에서는 각각의 약물에 대하여 자세하게 알아보도록 하겠다.
1. 항섬유화제 치료
IPF 환자를 대상으로 한 임상연구에서 피르페니돈(pirfenidone)과 닌테다닙(nintedanib)은 IPF에서 섬유화의 진행을 늦춰주고 사망률을 줄이는 효과를 보여주었다[8,9,40]. 피르페니돈은 형질전환성장인자-베타(transforming growth factor-ß)로 자극된 콜라겐 합성을 억제하고 세포외기질을 감소시키며, 섬유아세포의 증식을 차단하는 항섬유효과를 나타내는 약물이다. IPF 환자를 대상으로 한 ASCEND(Assessment of Pirfenidone to Confirm Efficacy and Safety in Idiopathic Pulmonary Fibrosis)연구에서는 FVC 예측치가 50% 이상인 중등증의 환자를 대상으로 피르페니돈 2,403 mg을 52주간 처방한 환자와 대조군을 비교했을 때, 피르페니돈 처방군에서 대조군에 비해 FVC의 감소와 사망을 유의하게 감소시키는 것을 확인할 수 있었다[9]. 분류가 불가능한 PF-ILD 환자를 대상으로 한 2상 임상연구에서도 IPF 환자와 마찬가지로 피르페니돈 2,403 mg을 24주간 복용한 환자군에서 대조군과 비교하여 FVC와 DLCO의 감소를 유의하게 감소시키는 것을 확인할 수 있어, IPF가 아닌 PF-ILD 환자에서도 피르페니돈이 효과가 있을 가능성을 보여주었다[16]. 외국에서 시행된 위의 연구들에 따르면, 피르페니돈의 투여용량은 267 mg 하루 세 번 복용으로 시작하고, 1주일 후 534 mg 하루 세 번으로 증량하며, 둘째 주 이후에 801 mg 하루 세 번 복용으로 증량하여 총 2,403 mg을 투여하도록 디자인되었다[9,16]. 이와 달리 2005년 일본에서 IPF 환자를 대상으로 한 무작위대조군연구에서는 피르페니돈 1,800 mg을 투여하였을 경우 또한 FVC 를 유의하게 감소시키고 운동능력을 유의하게 증가하는 효과를 보여주었다[41]. 이를 바탕으로 국내에서는 IPF 환자에게 1,800 mg의 피르페니돈을 투여하도록 권고하고 있다. 피르페니돈의 흔한 부작용은 오심, 소화불량, 식욕감소, 광과민성, 간수치 상승 등이 있으며, 이러한 부작용들은 보통 약을 복용하면서 점차 호전된다[9]. 이러한 부작용을 줄이기 위해 실제 임상에서 투약 시 처음에는 1일 3회 1정(200 mg)씩 복용을 하고, 2주 간격으로 매 회 1정씩 증량하여 4주 후에는 투여용량이 총 1,800 mg이 되도록 한다[42]. 부작용을 줄이기 위해 구토억제제나 장운동촉진제 등을 함께 사용할 수 있으며, 실외활동 시에는 자외선차단제를 사용하도록 권고한다. 또한 간기능 이상을 유발할 수 있으므로 투여 전과 투여 중 간기능 검사를 시행하여 이상유무를 확인하여야 한다. 국내에서는 2015년 10월부터 보험 급여로 피르페니돈의 처방이 가능해졌으며, 그 기준은 다음과 같다. FVC가 예측치의 90% 이하이거나 폐확산능의 예측치가 80% 이하인 경우, FVC가 예측치의 90%를 초과하고 폐확산능이 예측치의 80%를 초과하더라도 다음 중 두 가지 이상에 해당하는 경우는 보험급여가 가능하다. 1) FVC가 예측치의 연간 10% 이상 감소하거나 절대값이 200 mL 이상 감소하는 경우, 2) 임상증상 악화, 3) 흉부영상 악화 소견이 있는 경우이다. 하지만 현재 기준에 따르면 IPF를 제외한 다른 원인으로 인한 PF-ILD에 대해서는 피르페니돈을 급여로 처방할 수 없으므로 처방에 주의를 요한다.
다음으로 국내에서 처방 가능한 항섬유화제로는 닌테다닙이 있다. 닌테다닙은 섬유아세포 성장인자, 혈관내피세포 성장인자, 혈소판유래 성장인자의 세 가지 성장 인자를 동시에 억제하는 타이로신인산화효소(tyrosine kinases) 수용체 차단제이다[43]. IPF 환자를 대상으로 한 INPULSIS (Efficacy and Safety of Nintedanib in Idiopathic Pulmonary Fibrosis) 연구에서는 닌테다닙을 복용한 IPF 환자는 위약 투여 환자군 대비 연간 FVC의 감소가 약 50%까지 유의하게 줄었다[40]. 또한 하나의 2상 임상연구와 두 개의 3상 임상연구를 메타분석한 결과 닌테다닙을 복용한 환자에서는 FVC의 감소가 유의하게 감소되었을 뿐만 아니라 급성악화 위험을 47% 유의하게 감소시켰으며 세인트 조지 호흡기 설문(Saint-George’s Respiratory Questionnaire)으로 평가한 건강관련 삶의 질 감소 또한 유의하게 감소시키는 효과를 보여주었다[44]. PF-ILD 환자를 대상으로 닌테다닙의 효과를 보고자 한 3상 임상연구인 INBUILD (Nintedanib in Progressive Fibrosing Interstitial Lung Diseases) 연구에서도 닌테다닙의 복용은 위약 투여 대비 연간 FVC의 감소를 약 57% 지연시키는 것으로 나타나, PF-ILD 환자에서도 닌테다닙의 효과가 있음을 보여주었다[17]. 닌테다닙은 원칙적으로 1회 150 mg로 하루 2회 복용하도록 하며 피르페니돈과 달리 약제의 용량을 점차 증량할 필요는 없다. 가장 흔한 부작용으로는 설사가 있으며 그 외에 오심, 식욕감소 등이 나타날 수 있다[44]. 소화기계 부작용은 시간이 지날수록 점차 호전되며 부작용을 조절하기 위해 지사제나 항구토제 등을 사용할 수 있다. 부작용이 지속될 경우 용량을 100 mg 하루 2회로 감량하거나 약제 투여를 중단할 수 있으며, 피르페니돈으로의 교체 또한 고려해볼 수 있다[42]. 중등도 이상의 간부전이 있는 경우 투여의 금기이므로 투여 전후로 간기능 검사를 시행하여야 하며, 가임기 여성의 경우 닌테다닙 마지막 투여 후 3개월까지 피임이 필요하다[42]. 국내에서는 닌테다닙을 IPF, SSc-ILD 및 PF-ILD 환자에게 비급여로 처방할 수 있다.
2. 면역억제제 치료
IPF 환자에서는 면역억제제의 사용이 생존율 및 폐기능의 호전을 보이지 못하였으며, 오히려 사망률 및 약제 사용에 의한 부작용의 가능성을 높일 수 있어 현재는 사용을 하지 않도록 권고하고 있다[45]. 하지만 자가면역관련 ILD나 과민성폐렴 등 다른 원인에 의한 ILD의 경우 면역억제제의 사용이 권고되는 경우가 있다. 자가면역관련 ILD 중 유일하게 SSc-ILD에서 몇 개의 무작위대조군연구가 진행이 되었는데, 2006년에 발표된 Scleroderma Lung Study (SLS) I 연구에서는 cyclophosphamide (CYC) 투약이 위약군에 비해 FVC 감소 속도를 유의하게 늦추었으며, 약 49% 정도의 환자에서 FVC 호전을 보여주었다[46]. 하지만 CYC의 장기 사용은 악성질환의 발생 가능성을 높일 수 있고, 중단 1년 후에는 치료효과가 지속되지 않는다는 문제점이 있었다[47]. 다음으로 SSc-ILD의 치료에 대해 연구된 약물로는 mycophenolate mofetil (MMF)이 있다. MMF는 활성형인 mycophenolic acid의 전구약물로서, 체내에서 mycophenolic acid로 전환되어 선택적, 비경쟁적, 가역적으로 inosine monophosphate dehydrogenase를 차단하여 T림프구와 B림프구의 DNA와 RNA 합성을 저해하여 증식을 저해한다[48]. 여러 동물실험을 통해 MMF가 이러한 면역억제 효과 이외에도 평활근세포 및 섬유모세포의 분화를 억제하는 항섬유효과를 가지고 있음이 밝혀졌다[49]. 2016년 발표된 SLS II 연구에서는 MMF를 24개월 동안 투약한 환자군과 CYC를 12개월 동안 투약한 대조군을 비교하였고 양 군에서 FVC 감소 저하 효과는 차이가 없으나 MMF가 부작용이 적다고 보고하였다[50]. 2017년 SLS II의 MMF 투약군과 SLS I의 위약군을 비교한 연구에서는 MMF가 위약에 비해 24개월까지 지속되는 FVC, DLCO, 호흡곤란의 향상 효과를 보여주었다[51]. 이러한 연구결과를 바탕으로 2020년 10월부터, SSc-ILD 환자에서 한 종 이상의 타 면역억제제 투여에도 불충분한 반응을 보이면서, FVC가 45%에서 80% 사이인 경우 MMF를 급여로 투여할 수 있도록 급여기준이 확대 적용되었다.
결론
PF-ILD는 치료에 반응하지 않고 진행하는 섬유화를 보이는 여러 ILD를 하나의 임상적인 질병군으로 지칭하는 새로운 개념이다. 진행하는 폐섬유화를 가진 환자의 경우 이전까지는 사용할 수 있는 약물이 제한되어 있어 그 예후가 좋지 않았던 것이 사실이다. 기존에는 IPF에 대한 연구가 주로 이루어져 왔으나 최근 들어 PF-ILD에 대한 관심이 높아짐에 따라 항섬유화제 및 면역억제제의 PF-ILD에 대한 효과를 알아보기 위한 여러 연구들이 진행 중이다. IPF에 사용 중이던 피르페니돈 및 닌테다닙이 IPF가 아닌 PF-ILD에도 효과를 보여주어 그 적응증 또한 점차 확대되고 있으며, MMF도 SSc-ILD에서 좋은 효과를 보여주고 있다. 최근에는 폐섬유증을 억제하는 것을 넘어서 폐섬유증 자체를 줄이는 약물에 대한 연구도 활발하게 진행 중이다. 이러한 연구들이 성공적으로 진행된다면 폐섬유증 치료의 새로운 시대가 열릴 것을 기대해 본다.