서론
임상 진료 현장과 보건의료에서 매일 많은 양의 의료 정보가 발생되며, 이러한 의료 정보를 통한 연구결과들은 질환에 따른 위험성 예측과 치료의 방법을 좀 더 정확하게 선택하기 위한 기회를 제공한다. 최근의 의료 정보를 소위 빅데이터라고 지칭하며, 최근에는 여러 머신러닝 분석방법을 이용하여 빅데이터의 단순 관찰로부터 새로운 지식을 추출할 수 있다[1]. 의료 빅데이터로부터, 우리는 유전자들 간의 상호작용 및 유전자들과 주위 환경적 요인 간의 상호작용들이 각각의 객체에 어떤 영향을 미치는지를 알고자 하며, 생물정보학자(bioinformatician), 생물통계학자(biostatistician) 및 의학역학자(epidemiologist)들이 통계학적인 확률(statistical probabilities)로 임상 데이터 내에서, 유전자와 유전자 간의, 유전자와-환경적 요인들 간의 원인 인과관계(causal interactions)를 나타냈다[2]. 지금까지 많은 연구에서 의료 빅데이터를 분석하여 상호작용을 통계학적인 확률로 나타낼 때는 선형 회귀나 로지스틱 회귀와 같은 전통적인 통계 분석방법들을 사용하였다[3-5]. 임상 진료 시 발생되는 수많은 데이터와 각각의 객체 내에서의 유전자 발현 정보는 매우 복잡한 관계를 가지고 있으며, 우리는 이러한 수많은 인자들 간의 복잡한 관계에서 질환의 발병 및 진행에 관한 주요 인자들 간의 원인 인과관계를 알아야 질환의 치료 및 효과적인 질환 진행 억제를 이룰 수 있다. 회귀 분석을 이용한 기존의 통계 분석방법은 많은 연구자들이 광범위하게 사용하고 있어 비교적 쉽게 이용할 수 있으나, 각각의 분석 타겟(outcomes)에 따라서 각각의 다른 모델을 적용해야 하며, 비선형 관계의 분석에는 어려움이 있고, 무엇보다도 원인 인과관계를 효과적으로 모델링 하기에는 매우 제한적이다[1]. 베이시안 네트워크(Bayesian networks) 분석은 아직 널리 알려져 있지 않아 익숙하지 않으나, 하나의 모델로 여러 개의 타겟을 알 수 있으며, 무엇보다 원인 인과관계를 직관적으로 보여준다[2]. 따라서 베이시안 네트워크를 이용하여 머신러닝 분석방법을 통한 학습을 통해서 유전자 정보에서 알고자 하는 타겟 결과와 관련된 주요 유전자들의 조절 네트워크(gene regulatory network) 모델을 추출해 낼 수 있다[6].
본론
베이시안 네트워크 분석을 통한 확률적 그래픽 모델(probabilistic graphical model)은 기존의 통계분석에서 사용하는 빈도학파적 분석(frequentist analysis)과는 달리, 주어진 데이터 값을 가지는 모든 변수들의 원인관계를 수많은 그래픽 모델로 표현하며, 이중에서 현재의 데이터 값을 가장 잘 설명할 수 있는 가능성을 가지는 그래픽 모델을 제시하는 것이다[1]. 예를 들어 유방암의 골격계 전이암에 관련된 유전자들의 조절 네트워크 모델을 알기 위해서는 유방암의 골격계 전이암 환자들의 유전자 발현 정보를 얻고, 유방암은 있지만 골격계 전이가 없는 환자들의 유전자 발현 정보를 얻은 후에, 모든 유전자들의 발현 정보와 골격계 전이 여부와의 관계를 보여주는 모든 그래픽 모델을 추출한다. 이중에서 가장 현재의 데이터를 잘 설명할 수 있는 모델을 선택하는 분석방법이 베이시안 네트워크 분석방법이다. 이러한 분석 시 기존의 연구나, 통계학적 분석방법에서 의미 있는 변수들에 대해서 베이시안 네트워크 분석 시 더 가중치를 두어서 분석을 할 수 있으며, 주요 유전자들의 조절 네트워크 모델을 얻기 위해서 타겟 결과 변수와의 관련성(Markov blanket)을 이용하여 수만 개의 유전자들의 변수들을 점차 줄여서 주요 조절 유전자 몇 개와 타겟 결과 변수와의 관계로만 이루어진 베이시안 네트워크를 만들어낸다[6,7].
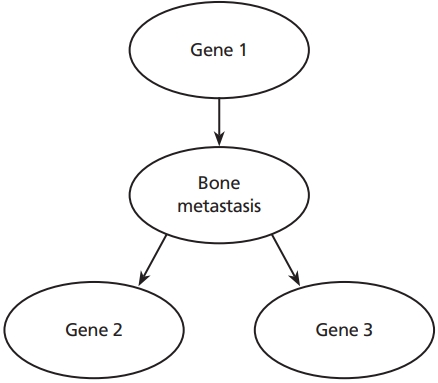
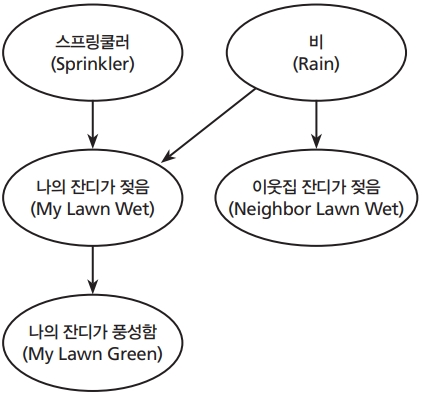
이러한 베이시안 네트워크 구조에 대해서 알아보면, 베이시안 네트워크는 방향성 비사이클 그래프(directed acyclic graph)이며, 각각의 변수들(variables)을 노드(node)라고 하며, 변수 사이의 관계는 아크(arc)로 표시하여 노드와 노드 사이의 인과관계가 있으면 화살표로 이어진다. 그래서 베이시안 네트워크에서, 아크는 원인 변수에 해당하는 부모 노드(parent node)와 직접적으로 영향을 받는 변수인 자녀 노드(child node) 사이의 관계를 표시한다(Figure 1). 이러한 베이시안 네트워크 구조는 마르코프 조건(Markov conditions)에 기준하여 형성된다[8]. 조건부 독립에 해당하는 마르코프 조건에 대한 이해는, 베이시안 네트워크의 구조 및 의미를 이해하기 위해서 필수적인 내용이므로 이어지는 그림으로 설명을 하려고 한다. Figure 2와 Figure 3은 조건부 독립에 대해서 설명을 할 때 많이 이용되는 그림들이다. Figure 2를 보면, 비가 오거나 나의 잔디에 있는 스프링쿨러를 작동시키면, 나의 잔디는 젖는다. 또한 비가 오면서 동시에 나의 잔디에 있는 스프링쿨러가 작동이 되어도 나의 잔디는 젖는다. 그리고 나의 잔디가 충분히 젖어 있으면, 나의 잔디는 풍성해질 것이다. 나의 잔디의 스프링쿨러만 작동을 하고 비가 오지 않으면, 나의 잔디는 젖고, 또한 풍성해질 것이다. 하지만 이러한 경우에 이웃집 잔디는 젖어 있지 않을 것이다. 이러한 네트워크는 Figure 3에서 3개의 하부 네트워크로 나누어 볼 수 있다. Figure 3A의 모양을 convergence arcs라고 부른다. 만일 우리가 잔디가 젖어 있음을 알고, 동시에 비가 오지 않았음을 알았다면, 나의 잔디에 있는 스프링쿨러가 작동될 경우(chance)가 매우 높다는 것을 의미한다. 즉 만일 노드 A (스프링쿨러)와 노드 B (비)가 노드 C (잔디 젖음)으로 convergence되면, 노드 C의 정보가 주어졌을 때, 노드 A와 노드 B는 의존적인 관계가 된다. 즉 나의 잔디가 젖어 있을 때, 그 원인이 나의 잔디의 스프링쿨러가 작동되거나 혹은 비가 올 경우 두 가지 가능성 밖에 없기 때문에, 스프링쿨러의 작동 가능성이 높아지면, 비가 왔을 가능성은 낮아지고, 스프링쿨러의 작동 가능성이 낮아지면, 비가 왔을 가능성이 높아진다. 그러므로 convergence에서는 child node의 정보를 알 경우에, parent node들의 관계는 서로에게 영향을 미친다. Figure 3B의 모양은 divergence arcs라고 부른다. 이때 만일 당신이 비가 온 것을 안다고 했을 때, 나의 잔디가 젖어 있다는 정보가, 이웃집 잔디가 젖어 있는 가능성에 영향을 미치지 않는다. 비가 온 것에 대한 정보는 이웃집 잔디가 젖어 있는 가능성에 영향을 미치지만, 비가 온 것에 대한 정보를 알고 있을 때 내 잔디의 젖어 있는지 여부가, 이웃집 잔디가 젖어 있는지 여부에 대해서 영향을 미치지 못한다. 즉 divergence arcs에서는 node A (비)를 알고 있을 경우에 node B (나의 잔디가 젖음)와 node C (이웃집 잔디가 젖음)와의 관계는 상호 독립적이다. Figure 3C에서의 관계를 serial arcs라고 부른다. 만일 나의 잔디가 젖어 있으면, 스프링쿨러의 작동 여부가 잔디의 풍성함에 더 많은 영향을 미칠지 여부를 알 수 없다. 즉 serial arcs에서는 변수 A (스프링쿨러 작동)에서 변수 B (나의 잔디 젖음)로, 변수 B에서 변수 C (나의 잔디가 풍성함)로 이어지며, 변수 B의 정보가 주어지면, 변수 A와 변수 C는 서로 독립적인 관계이다. 베이시안 네트워크는 이 세 가지 형태(converging, diverging, serial)로 구성되어 있으며, 이러한 관계 이해는, 최종적으로 얻어진 베이시안 네트워크의 형태 해석에 필수적이다.
이제 베이시안 네트워크를 이용하여 유전자 조절 네트워크 모델을 추출하는 연구방법에 대한 예시과정을 보여줌으로써, 베이시안 네트워크에 대한 이해를 한층 더 높이고자 한다. 저자는 폐암의 골 전이에 관련되는 주요 유전자들의 조절 네트워크를 얻고자 하였다.
1. 데이터 수집 및 데이터 정제
유전자 발현 마이크로 어레이 정보를 얻고자 National Center for Biotechnology Information, NCBI에서 제공하는 Gene Express Omnibus (https://www.ncbi.nlm.nih.gov/geo/)를 통해서 인체 폐암과 폐암의 골 전이 조직에서 얻어진 유전자 정보를 수집하였다[9]. Gene Express Omnibus에서 총 3개의 관련 연구들(GSE76194, 29391, 32474)을 확인할 수 있었으며, 3개의 연구들에서 20명의 환자 유전자 발현 정보를 다운로드 받았다. 발현 정보 변수는 연속형 값을 가지고 있어서, z-score 표준화 방법을 이용하여, 범주형 변수로 변경하여, 저 발현, 정상 발현 및 고 발현의 세 가지 범주로 분류하였다. 20명의 환자들의 발현 총 유전자들 중에서 17,260개의 유전자들이 공통으로 발현되었으며, 이 중에서 골 전이 유무와 발현 변화의 상관관계의 수치가 상위 10%에 해당하는 유전자인 1,726을 추출하였으며, 기존의 연구 논문들에서 언급된 폐암의 골 전이와 밀접한 관련이 있는 74개의 유전자들도 추가하여 총 1,788개의 유전자들을 선택하였다[10-12].
2. 베이시안 네트워크 분석 및 후보 유전자들의 선택
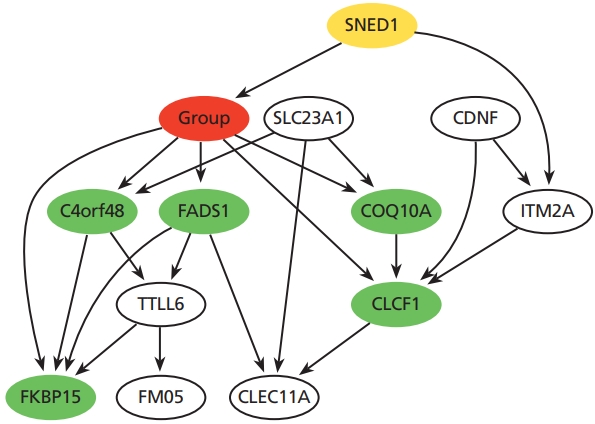
베이시안 네트워크를 얻기 위해서 Bayesian Network Inference with Java Objects 프로그램을 이용하여 분석을 하였다[7,13]. 여러 번의 베이시안 네트워크 분석 및 Markov blanket 내의 관계를 이용하여 주요 유전자 조절 네트워크를 만들었다(Figure 4). 1,788개의 유전자를 24개의 유전자로 줄이기 위해서, 3번의 축소(downsizing)를 거쳤으며, 매번 축소과정에서 타겟 결과인 폐암의 골 전이 노드와 1차적으로 아크를 가지는 노드를 선택하였다. 또한 3번의 베이시안 네트워크 분석 중에서 최적의 네트워크를 얻기 위해서 분석시간은 3시간, 6시간, 12시간 24시간, 48시간씩 각각 3번 반복 분석하였으며, 각각의 분석시간 및 횟수에서 최적의 가능성을 보여주는 네트워크를 선택하였다. Figure 4에서 보여주는 베이시안 네트워크를 보면, SNED1 유전자의 발현 여부가 폐암의 골 전이에 원인 유전자관계로 있으며, 그 밖의 녹색으로 채워진 노드들의 유전자들은 폐암이 골격계 전이된 이후에 영향을 받는 유전자들임을 알 수 있다.
3. 베이시안 매개변수(parameter)들의 학습
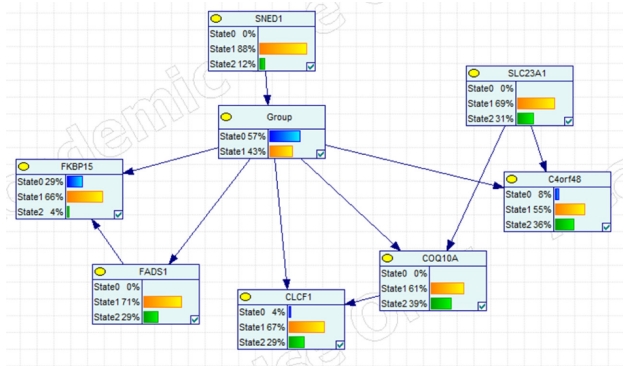
현재의 데이터를 가장 잘 설명하는 최종 베이시안 네트워크 모델을 얻었으면, 이후에 이 모델의 매개변수들이 어떠한 확률값을 가지고 상호작용을 하는 것을 알 수 있는 그래픽 모델을 GeNIe (BN Graphical Network Interface; BayesFusion, LLC, Pittsburgh, PA, USA) 프로그램을 이용하여 얻을 수 있다(Figure 5). Figure 5를 보면, 현재 데이터는 골 전이가 없는 폐암 환자의 비율은 57%이고, 골 전이가 있는 환자의 폐암 비율은 43%인 데이터에서 SNED1 유전자의 발현은 정상 혹은 과발현되어 있으며, 그 밖의 자녀 노드의 발현은, FKBP15 유전자를 제외하고, 나머지 노드들의 유전자 발현은 정상 혹은 과별현의 경향을 가진다.
이상으로 간략하게 베이시안 네트워크의 기본 개념 및 구조와 실제로 베이시안 네트워크를 이용하여 주요 유전자 조절 네트워크를 얻어내는 방법에 대해서 소개하였다.