서론
차세대 염기서열 분석법(next-generation sequencing, NGS)의 발전으로 현재 약 100만원 정도의 비용이면 10일 내외로 개인의 유전체 변이를 검사하는 것이 가능하게 되었다. 향후 수년 이내에는 인간의 30억 쌍의 전체 염기서열을 100달러 미만의 가격으로, 과거에는 상상하지 못했던 매우 저렴한 비용으로 검사할 수 있게 될 것으로 예상되고 있다[1]. 이에 발맞춰 우리나라에서는 2017년 3월부터 고형암과 유전 질환을 대상으로 유전자 패널검사를 시행할 경우, 본인 부담 50%의 선별 급여가 적용되고 있다. 2023년 3월 기준으로 건강보험심사평가원에 유전자 패널검사 실시 기관으로 승인을 받은 곳은 69개소에 이른다. NGS 검사가 급여권으로 들어오며, 많은 병원의 진료 현장과 임상 검사실에서도 NGS를 이용한 다수의 유전 질환, 그리고 고형 및 혈액암에 대한 유전자 검사가 진행되고 있다[2-4]. 최근에는 국가 바이오 빅데이터 시범사업, 개별 연구자의 임상연구, 유전체 회사를 통한 유전자 검사 시행 등 다양한 경로를 통하여 유전체 데이터가 생산되고 있어, 이에 대한 적절한 활용과 논의가 중요해진 상황이다.
국내의 경우, 식약처와 관련 학회들에서 다양한 지침과 가이드라인들을 발표하여 유전자 검사의 시행과 해석에 대한 표준화를 위하여 노력하고 있다[5-7]. 그럼에도 불구하고, 유전자 검사 결과를 받아보는 환자와 임상 의사의 입장에서는 유전체 데이터로부터 검출된 다양한 변이에 대하여 적절하게 해석하고, 필요한 경우에는 가족 검사 등의 추가적인 검사를 시행하거나, 유전 상담을 포함하는 진료 활동에 이를 적절하게 활용하는 방법에 대한 이해가 부족한 상황이다. 더불어 유전자 검사는 검사법의 한계와 그 의미를 명확히 이해한 상태에서 환자에게 충분한 동의를 받고 시행하도록 권장되고 있으나[8], 아직까지 이러한 절차에 대한 사회적 합의와 규범적 준거 또한 미흡하다.
임상 검사실에서 가장 흔하게 시행하고 있는 엑솜(exome) 또는 타겟 패널(target panel) 염기서열분석 방법은 인간의 전체 유전체 중 약 1-2%에 해당하는 단백질 부호화 서열 또는 그 중 일부에 대한 변이 정보만을 확인할 수 있다. 이러한 검사의 한계에도 불구하고, 희귀 변이와 질병과의 연관성이 잘 알려진 신경계, 심혈관계, 근골격계, 가족성 암 등과 같은 다양한 유전성 질환에서는 엑솜 염기서열분석의 임상적 유용성이 다수 보고된 바 있다[9,10]. 또한 모든 유전체 서열을 검사하는 전장 유전체(genome) 염기서열분석 방법은 비용과 검사 판독 소요 시간이 더 길지만 더 높은 진단율을 달성할 수 있음이 보고되어, 점차 활용 범위가 확대될 것으로 예상되고 있다[11,12]. 그러나 최근에는 유전자 검사에 대한 적절한 이해없이 마구잡이로 시행되는 검사가 늘어나면서, 그 결과로 발견되는 변이들의 의미를 해석하기 위해 환자들이 여러 병원을 순회하는 ‘유전자 변이 해석 방랑’ 사례 또한 점점 늘어나고 있다.
이에 이 논문에서는 NGS 시대에 임상 진료 환경에서 자주 마주치게 되는 유전자 검사 결과에 대한 적절한 해석과 추가적인 가족 검사를 활용하기 위한 적절한 지침과 사례, 그리고 고려해야 할 사항들에 대해 안내하고자 한다. 이를 통하여 많은 임상의들이 NGS 시대의 유전자 검사에 대한 적절한 활용법과 한계점을 이해하고, 동시에 유전자 검사의 해석, 가족 검사, 그리고 유전 상담 과정에서 마주치게 되는 많은 기술적 및 윤리적 문제들에 대해서 생각해볼 수 있기를 바란다.
변이 검출의 기술적 측면과 한계
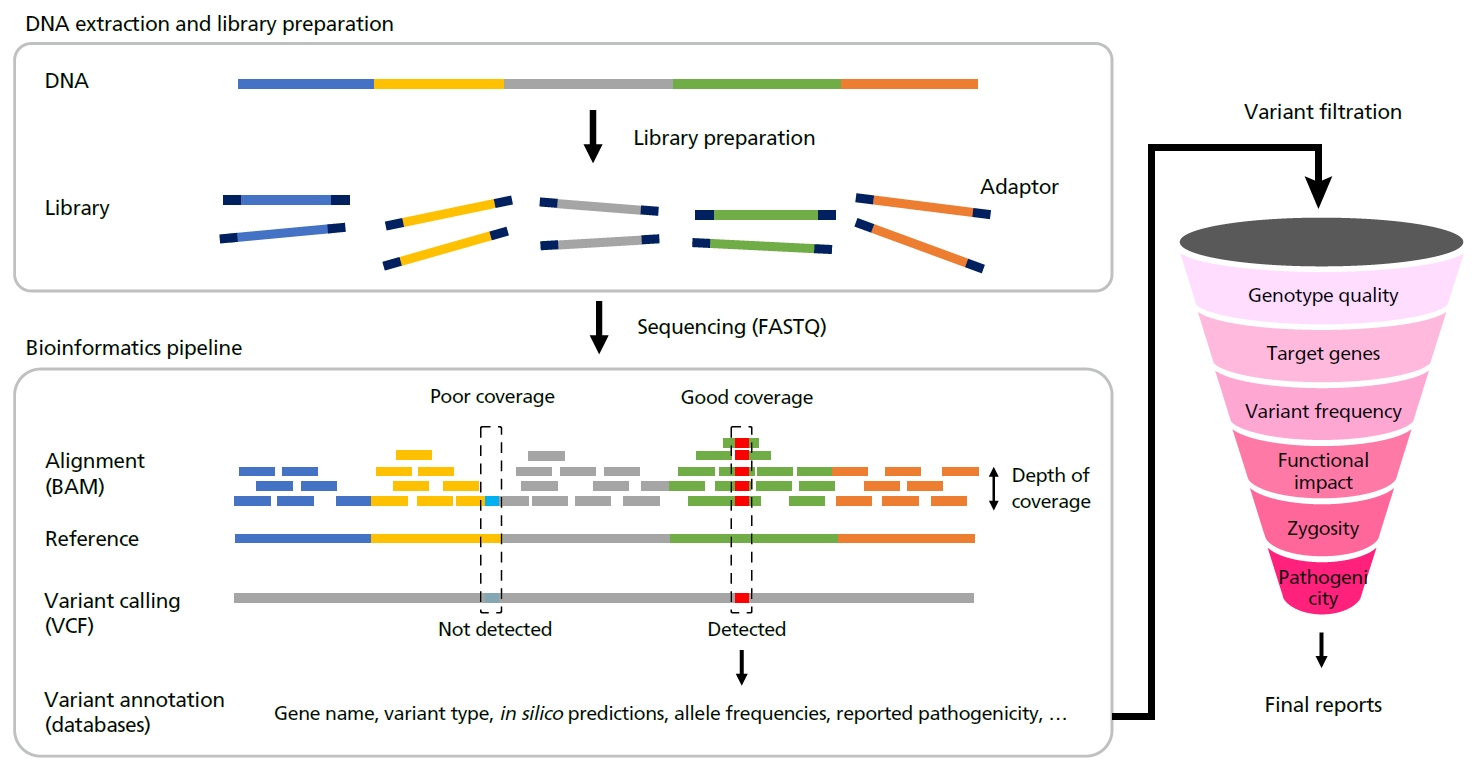
NGS를 이용한 유전자 변이 검출 원리에 대해 간단히 소개하고자 한다. 현재 임상 검사실에서 가장 널리 사용되고 있는 염기서열분석 방법은 짧은 리드(short-read) 방법이다. 짧은 리드 염기서열분석을 하기 위해서는 DNA 도서관 (DNA library)을 제작해야 하며, 환자에서 채취한 검체에서 추출한 DNA를 500개 염기서열 정도의 작은 단편들로 조각 낸 후 어댑터를 붙이게 된다(Figure 1). 이렇게 제작된 DNA 도서관을 이용하여 염기서열분석기기에서는 약 100-200 염기서열 정도의 단위로 수백만 개 이상의 리드들을 동시에 읽어 들이게 된다. 이렇게 생성된 리드들은 조각난 DNA 서열의 정보를 포함하고 있기 때문에, 컴퓨터 알고리즘을 이용하여 인간의 참조 유전체 서열에 정렬하여 재구성한다.
따라서 짧은 리드 염기서열분석 방법은 DNA를 조각내고 재구성하는 과정에서 정보의 소실이 발생할 여지가 있다. 리드 자체에 발생하는 오류, 리드 정렬의 정확도, 그리고 해당 변이를 커버하는 리드의 개수 등에 따라 민감도와 특이도, 그리고 정확도가 달라질 수 있음을 이해하는 것이 필요하다[13]. 유전체 영역의 구아닌 또는 싸이토신 염기 비율(guanine-cytosine 분율)이 높거나 동일 서열이 반복되는 부위에서는 리드가 적절하게 생성되지 못하거나 제대로 정렬하지 못하게 되어, 위음성이 발생할 확률도 존재한다. 더불어 짧은 리드 염기서열분석 방법은 삼염기 반복질환에서 흔하게 나타나는 짧은 반복수 변이나 다양하고 복잡한 구조 변이의 검출에는 한계가 있다.
NGS 기반의 유전자 검사는 그 임상적 유용성으로 인하여 다양한 변이를 동시에 검출하는 가장 효율적인 검사법으로 자리매김하고 있다. 그러나 앞서 언급한 기술적 한계 이외에도 검체의 전처리, 생산된 데이터의 보관 및 처리를 위한 파이프라인의 자동화 및 표준화, 그리고 정확도와 재현성 등 함께 해결해야 할 다양한 이슈들이 존재한다[14]. 최근에는 짧은 리드 방법의 한계점을 극복하고 더 정확하게 변이를 검출을 하기 위하여, 긴 리드(long-read) 염기서열분석 등의 다양한 기술들이 개발되고 있으며, 검사 비용의 문턱이 점점 낮아지게 된다면, 임상 검사실에서도 이러한 방법들이 점차 널리 쓰이게 될 것으로 예상된다.
변이의 판독 과정
국내 NGS 검사를 통하여 검출된 변이들은 일반적으로 미국 의학유전학회 및 분자병리학회(American College of Medical Genetics and Genomics/Association for Molecular Pathology, ACMG/AMP)에서 권장하는 가이드라인에 따라 변이의 임상적 중요도를 5개의 카테고리로 분류하고, 병적으로 예측되는 변이들을 검사 결과지에 보고하도록 하고 있다[6]. 다만 이러한 기준은 생식 세포(germline) 변이를 대상으로 유전형과 질환과의 관련성이 명확하게 수립된 멘델 유전 질환에만 적용할 수 있으며, 체세포(somatic) 변이를 대상으로 하는 암종이나 다인자 유전 질환에는 적용하기가 적절하지 않다. 한 개인에서 검출되는 변이의 개수는 인종, 패널의 종류, 리드의 생산량, 검사의 퀄리티 등에 따라서도 다양한 편차가 존재하며, 엑솜 염기서열분석의 경우 대략 수십만개 이상의 변이를 검출하게 된다. 따라서 이 중에서 질병과 관련된 원인 변이를 찾기 위해서는 여러가지 기준에 따라서 변이를 적절하게 필터링하여, 가장 가능성이 높은 후보를 찾고, 최종적으로 환자의 임상 증상과 잘 맞는 변이를 찾는 과정이 요구된다. ACMG/AMP 가이드라인에서는 인구 집단 내 변이 빈도, 컴퓨터 도구를 이용한 변이의 기능 예측, 변이의 실험적 근거, 가계도 상에서 변이의 기원과 유전 양식, 드노보(de novo) 변이 여부, 기보고된 데이터베이스 등을 활용하여, 근거 수준의 중요도와 조합에 따라 변이를 카테고리별로 분류하게 된다[15]. 아래에서는 ACMG/AMP 가이드라인에서 고려하는 변이 분류의 주요 개념들과 활용할 수 있는 데이터베이스들에 대해 설명하겠다.
1. 변이 빈도와 유전 질환의 유병률
특정 변이가 유전 질환의 원인 변이이고 투과도가 100%라고 가정한다면, 해당 변이를 가진 사람들은 모두 유전 질환에 이환될 것이기 때문에, 해당 변이의 인구 집단에서의 빈도는 유전 질환의 유병률보다 높을 수 없다. 따라서 우리는 이러한 단순한 가정을 통하여 변이의 인구 집단에서의 빈도를 기준으로 하여, 중요도가 떨어지는 변이들을 필터링할 수 있다. 그러나 실제로 이러한 관계는 형질과 유전 변이와의 관계를 나타내는 유전적 조성이나 유전 양상에 따라서 조금 더 복잡하게 영향을 받게 되는데, 가령 상염색체 우성인지 또는 상염색체 열성인지에 따라서, 해당 변이를 하나의 염색체에만 가지고 있어도 되는지 또는 양쪽 모두에 가져야 하는지가 달라지게 된다. 따라서 일반적으로 상염색체 우성 유전 질환의 경우에는 훨씬 더 낮은 변이 빈도를 기준으로 필터링을 하게 되며, 상염색체 열성 질환의 경우에는 정상 보인자들이 인구 집단 내에 어느 정도 존재하기 때문에, 조금 더 느슨한 빈도를 기준으로 적용하게 된다. 이에 따라 일반적으로 상염색체 우성 질환은 0.1% 미만, 상염색체 열성 질환은 1% 미만의 변이 빈도를 갖는 변이로 우선적으로 필터링을 해볼 수 있다[16]. 다만 변이의 빈도는 모집단의 특성과 크기에 따라서 달라질 수 있는 상대적인 값이므로, 절대적인 기준이 있는 것은 아니다. 특히, 매우 낮은 비율로 존재하는 희귀 변이는 유전적 선택압의 결과로 인종과 유전적 조성에 따라 달라질 수 있기 때문에[17], 한국인을 대상으로 하는 대규모 변이 빈도 데이터베이스를 구축하고 활용하는 것이 매우 중요하다. 현재까지 한국인을 포함하는 변이 빈도에 대한 다양한 데이터베이스가 구축되어 있으며, 대표적으로 gnomAD, KRGDB, Korea1K, KOVA2 등이 있다(Table 1) [18-29].
2. 변이의 종류와 생물학적 결과
유전자의 변이는 삼염기 유전자부호(codon)의 해독 과정에 영향을 미쳐, 다양한 생물학적 결과를 초래할 수 있다. 가령 단백질 합성의 시작을 일찍 멈추게 하거나, 해독의 틀을 옮겨서 잘못된 단백질이 만들어지게 하거나, 잘라이음(splicing) 과정에 영향을 미치거나, 원래의 아미노산을 다른 아미노산으로 치환시키거나, 아무런 영향이 없을 수도 있다. 따라서 변이가 유전자의 어느 부위에 존재하며, 어떠한 기능적 결과를 미치는지 구분하는 것은 변이의 해석 과정에서 매우 중요한 단계이다. 특히, 아미노산이 치환되거나 잘라이음 과정에 영향을 미칠 것으로 생각되는 변이들은 다양한 기능적 결과를 초래할 수 있어, 많은 예측 도구들과 데이터베이스들이 개발되어 있으며(Table 1) [18-29], 변이들을 분류하는 과정에서 보조적인 근거로 사용될 수 있다[22,23].
3. 변이의 기원과 유전 양식
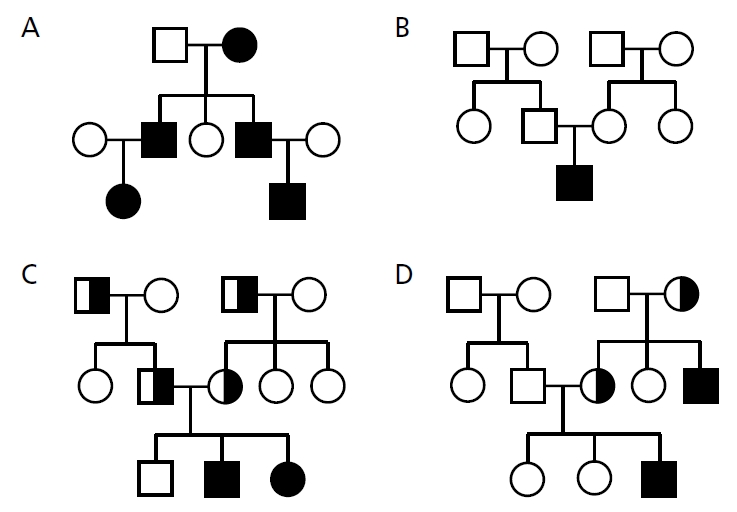
가계도로부터 유전 질환의 유전 양상을 추정하는 것은 변이를 해석하는 데 매우 중요한 정보를 제공한다(Figure 2). 일반적으로 염색체는 양쪽 부모로부터 하나씩 물려받게 되므로, 염색체 상에 존재하는 변이의 기원을 알게 되면, 유전 변이를 판독하는 데 활용할 수 있다. 예를 들어, 아버지와 아들이 환자라고 한다면, 원인 변이는 아버지와 아들이 공유하는 변이여야 하며, 아들이 가지고 있는 변이들 중 어머니에게 물려받은 대략 절반의 변이들은 모두 제외시킬 수 있다. 비슷한 원리로 하나의 큰 가계도 상에서 다수의 환자들이 있는 경우, 모두가 공유하는 변이로 원인 변이를 추려볼 수 있다(Figure 2A). 환자의 양쪽 부모가 모두 유전 질환에 이환되지 않은 경우에는 상염색체 우성 질환에서 환자에서 새롭게 발생한 드노보 변이를 원인으로 생각해볼 수 있으며(Figure 2B), 상염색체 열성 질환에 대해서는 양쪽 부모로 부터 하나씩 변이를 물려받아서, 우연히 특정 유전자에 대해 동형접합을 가지거나, 복합 이형접합을 가지는 조합이 원인 유전 질환이라고 추정할 수 있다(Figure 2C). 특히, 드노보 변이의 경우에는 일반적으로 전체 엑솜 상에서는 1-5개 정도만 발생하기 때문에, 기능적인 영향이 큰 변이라면 질환의 원인 변이일 우도(likelihood)가 매우 높은 변이로 생각될 수 있다. 이 경우에는 환자의 생물학적 부모에서 해당 변이 유무를 검사하여 드보노 여부를 평가하는 것이 매우 중요하다. 이렇듯 변이의 해석은 변이의 기원과 가계도, 그리고 유전 양식에 따라서 서로 다르게 해석될 수 있기 때문에, 임상의는 일차적으로 임상 양상과 가족력을 근거로, 후보 질환을 추정하고 유전 양식에 따라 감별 진단을 하는 것이 중요하다.
4. 변이 보고 데이터베이스
형질과의 관련성이 잘 알려진 변이들의 경우에는 기존의 보고를 근거로 하여, 변이를 직접적으로 분류할 수 있다. 따라서 이러한 정보들을 잘 분류하여 데이터베이스로 구축하는 것이 중요하며, 형질 또는 질환에 따른 카테고리 별로 다양한 데이터베이스들이 공개되어 있다. 가장 널리 알려져 있는 유전 질환의 변이 분류 데이터베이스로는 ClinVar [24]와 Human Gene Mutation Database [25]가 있으며, 약물 유전자 변이 정보를 포함하는 PharmGKB [26], 그리고 암의 변이 정보를 포함하는 Catalogue of Somatic Mutations in Cancer [27] 등도 임상적으로 널리 활용되고 있다(Table 1) [18-29].
유전 검사의 임상적 해석 및 활용
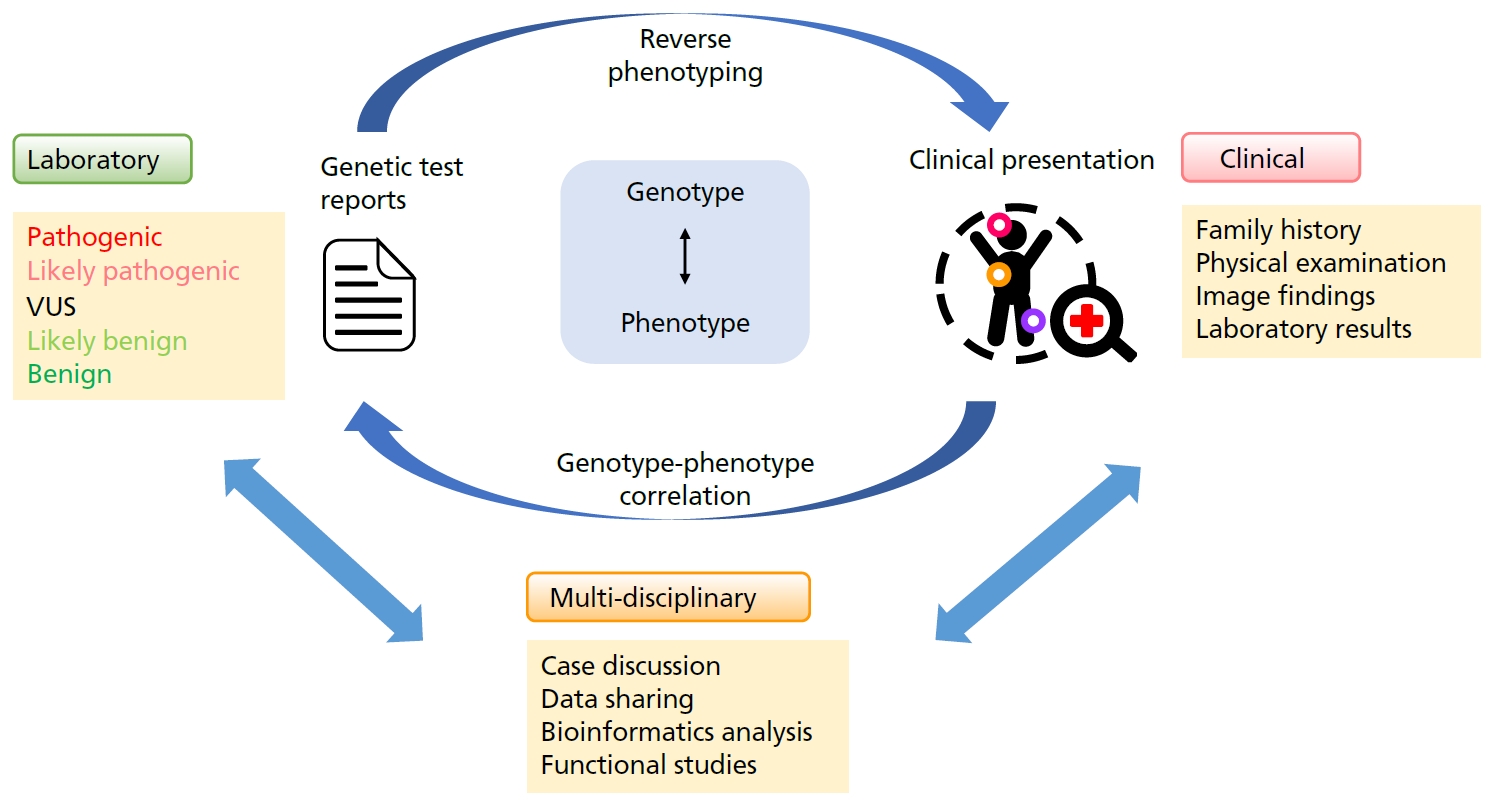
유전자 검사 결과지에서 병적(pathogenic) 변이 또는 유사병적(likely pathogenic) 변이라고 분류되었더라도, 유전 질환의 원인 변이라고 단정지을 수는 없다. 반대로, 의미가 불 분명한 변이(variant of uncertain significance, VUS)로 구분되었더라도 유전 질환을 일으키는 원인 변이일 수 있다. 변이의 분류는 우선적으로 중요도가 높은 유전 변이를 구분하기 위한 분류 체계로, 절대적인 기준이 아니며 근거 수준과 상황에 따라 바뀔 수 있기 때문에, 환자의 기왕력, 가족력, 신체 진찰 소견, 영상의학적 소견, 그리고 진단검사 결과 등과 함께 종합적으로 판단하는 것이 중요하다. 유전 변이와 환자의 임상적 소견을 서로 맞춰보는 과정을 유전형-표현형 대응(genotype-phenotype correlation)이라고 하며, 해당 유전 질환에 대한 의사의 경험과 안목이 중요한 과정이다(Figure 3). 최종 진단은 유전 변이가 확인된 유전자에서 알려진 임상적 특징과 환자의 임상 소견이 서로 일치하는 경우만 가능하며, 유전자 검사 결과지에만 의존하여 결론을 내릴 경우에는 잘못된 결론을 내릴 수 있어 주의가 요구된다[30].
일반적으로 유전형-표현형 대응 과정은 Online Mendelian Inheritance in Man [28] 또는 GeneReview [29]와 같이 유전자에 따른 표현형이 정리된 데이터베이스와 기보고된 문헌 등을 참조하여 검토해볼 수 있다. 환자에서 검출된 변이에서 알려져 있는 임상적 특징과 실제 환자의 임상 양상, 그리고 표현형이 얼마나 일치하는지 평가하고, 만약 환자의 임상 양상이 알려져 있는 내용과 일치하지 않는 경우에는 다음과 같은 가능성들을 고려해야 한다. 즉, 환자의 나이가 아직 질병이 발병하지 않는 시기이거나, 임상 양상을 아직 확인하지 못하여 추가적인 평가가 필요하거나, 잘못된 판독으로 유전형이 표현형과 관련성이 없는 경우 등이다. 반대로, 유전자 검사 결과에서 전혀 예상하지 못했던 유전자에서 매우 잘 알려진 병적 변이가 보고된 경우에는 환자에게서 고려하지 못했던 임상 증상이 없는지를 확인하는 과정이 필요하다. 이러한 과정을 역표현형 평가(reverse phenotyping) 과정이라고 말한다. 역표현형 평가는 변이의 해석과 유전 질환의 진단율에도 큰 영향을 미치는 중요한 과정으로[31], 해당 과정을 통하여 검출된 유전 변이의 임상적 의미를 평가하고 환자의 임상 양상을 재평가하게 된다. 이러한 전반적인 과정에는 임상의사, 유전 질환 전문의, 진단검사의학과 전문의, 그리고 생명정보학 전문가 등 다양한 배경을 가진 전문가들의 다학제적 접근(multidisciplinary team approach)이 중요하며, 이를 통해 전반적인 진단율을 향상시킬 수 있음이 알려져 있다[32,33].
경우에 따라서는 환자의 임상 양상이 유전 질환의 특성과 일부만 일치하거나, 알려져 있지 않은 새로운 표현형이 관찰될 수도 있다. 이러한 경우에는 유전자 진단이 잘못되었을 가능성, 기존에 알려져 있지 않은 새로운 표현형 확장, 그리고 추가적인 원인 변이의 존재 등을 모두 고려해봐야 한다. 잘못된 진단의 가능성을 배제하기 위해서는 기존 변이에 대한 재평가 및 역표현형 평가 과정이 중요하다. 최근에는 둘 이상의 병적 변이가 한 환자에서 나타나는 경우도 있어, 이러한 현상이 다양한 임상적 양상과 표현형 확장에 기여한다고 알려져 있다[34,35]. 따라서 이미 특정 유전자의 이상으로 결론이 내려진 환자라고 하더라도, 기존에 알려져 있는 유전자로 설명이 되지 않는 추가적인 임상 양상이 보이는 경우에는 추가적인 유전적 원인이 없는지 평가하는 과정이 필요하다. 특히, 임상 검사실에서 검사할 수 있는 범위를 넘어서는 추가적인 실험 또는 연구가 필요한 경우도 종종 있으며, 이 때문에 변이의 재평가 및 추가적인 유전 변이의 해석 과정은 다학제적 접근이 더욱 요구된다. 이러한 과정을 통하여 잘 알려져 있지 않았던 유전형과 환자의 임상 양상을 다양한 방면에서 이해하게 되며, 이를 위해서는 임상 검사실과 생명정보학, 그리고 중개 연구실의 역량과 각 분야의 유기적인 협력과 소통이 매우 중요하다고 할 수 있겠다.
가족 검사와 유전 상담
1. 가족 검사
위와 같은 과정을 통하여 유전자 검사 결과와 환자의 임상 양상을 평가하여도, 의미가 불분명하여 추가적인 평가가 필요한 유전 변이들이 다수 존재한다. 이러한 경우에 유전 변이의 기능을 직접적으로 측정하는 실험이 가장 확실하게 도움이 될 수 있으나, 많은 시간과 비용이 소모되어 현실적으로는 어려운 경우가 많다. 따라서 일반적으로는 가족 검사가 변이의 의미를 해석하는 데 가장 유용한 정보를 제공한다.
가족 검사란, 환자와 가족 관계에 있는 가족 구성원들의 유전형을 검사하는 것을 말하지만, 대부분의 경우는 부모의 특정 유전형을 검사하는 것을 일컫는다. 가족 검사는 환자에서 찾은 특정 유전형에 대해서, 부모의 검체를 채취하여 생어(Sanger) 염기서열분석을 통해 확인하는 것이 일반적이지만, 환자와 부모들의 검체를 처음부터 함께 수집하여 한꺼번에 엑솜 염기서열분석을 진행하거나 형제까지 포함하여 분석하는 형태로 진행할 수도 있다. 이러한 방법은 드노보 변이를 검출하거나, 가족 구성원 간에 공유하는 변이를 한 번에 확인할 수 있어서, 진단율을 향상시키는데 기여할 수 있다[36,37]. 예를 들어, 양쪽 부모가 정상이면서 상염색체 우성 질환이 의심되는 경우에는 드노보 변이를 우선적으로 검토해볼 수 있으며, 가계 내에서 다수의 환자가 존재하는 경우에는 환자들이 모두 공유하는 변이에 집중해볼 수 있다. 상염색체 열성 질환의 경우에는 부모가 각각 하나의 변이를 이형접합으로 가진 보인자여야하고 환자는 동형접합 또는 2개의 이형 접합 변이를 모두 가져야 하므로, 부모 검사를 통해 의심되는 변이가 각각 부모로부터 하나씩 물려받았는지를 확인하게 된다. 만약 2개의 변이가 한쪽 부모에서 관찰된다면, 해당 변이는 상염색체 열성 질환의 유전 양상에 합당하지 않으므로, 원인이 아니라고 결론 내릴 수 있다. 비슷하게, X-연관 열성 유전 질환의 경우에는, 어머니에게서 가족 검사를 시행하여, 보인자 여부를 확인할 수 있다. 이렇게 가족들의 유전형을 검사하여, 추가로 얻어진 근거들은 변이의 판독 결과에 영향을 미치게 되는데, 종종 의미가 불분명하다고 분류된 변이(VUS)를 유사 병적 변이로 재분류하기도 한다[38].
2. 유전 상담
유전 상담이란 유전 질환이 환자와 가족에게 어떠한 영향을 미칠 수 있는지에 대한 종합적인 정보를 제공하는 일련의 과정으로, 유전자 검사 이전과 이후로 구분할 수 있다. 검사 전 유전 상담은 유전자 검사의 의미와 한계, 예상되는 결과에 따른 계획 등에 대한 충분한 설명을 통하여 동의를 얻는 과정이며, 검사 이후에는 유전자 검사 결과에 대한 의미와 유전 진단이 내려진 경우 환자의 치료 방침 및 예후 등에 대한 안내와 해당 변이를 공유하고 있을 가능성이 높은 가족 구성원들에 대한 추가적인 상담을 진행한다[39]. 환자에서 확인된 유전 질환의 유전 양식에 따라 위험도를 예측할 수 있는데, 일반적으로 상염색체 우성 질환의 경우에는 자녀에게 해당 질환을 물려줄 가능성이 50%이며, X-연관 열성 질환의 경우에는 보인자에서 남아를 출생할 경우 질환이 발생하게 되고, 상염색체 열성 질환의 경우에는 부모가 다음 자녀를 계획할 때, 25%의 확률로 동일한 질환의 환자가 태어날 수 있다. 이를 고려하여 필요시에는 주변 가족 구성원들의 유전형을 단계별로 검사하고, 질환 이환의 위험도를 예측 또는 평가한다. 이를 통해, 다음 자손에게 질환을 물려줄 가능성에 대해서도 유전 상담을 진행할 수 있다(Figure 2).
유전 상담 과정에서는 동일한 유전 변이라 하더라도 개인별로 변이의 투과도가 다르게 나타날 수 있음을 고려해야 한다. 투과도란 해당 변이를 가지고 있을 때 질환에 이환될 확률을 나타내며, 유전 질환과 변이의 종류에 따라 다양한 투과도를 가질 수 있다. 더불어, 투과도가 100%라고 하더라도 임상 양상이나 중증도도 개인별로 서로 다르게 나타날 수 있다. 최근에는 체외 수정 시술의 발전으로 중증도가 심각한 200여개 유전 질환에 대해서는 원인 변이를 착상 전에 검사하여, 해당 변이를 갖지 않는 배아를 임신하는 착상 전 유전 진단(preimplantation genetic diagnosis)이 가능해지게 되었다(Table 2) [40,41]. 비슷하게 임신 중에도 비침습적 산전 유전 검사(non-invasive prenatal testing)를 진행하여, 융모막 또는 양수로부터 태아의 유전 질환 이환 여부를 검사할 수 있다. 다만 이러한 방법은 유전형과 유전 질환과의 관계가 명확하게 확립된 경우에만 가능하므로, 이환된 환자의 유전적 원인을 확실하게 찾는 선행 과정이 매우 중요하다. 원인 유전자와 변이를 찾은 경우에는 유전 질환의 유전 양식을 고려하여, 임신을 계획 중인 다른 가족 구성원에서 가족 검사를 권유하여 정확한 유전적 위험도 평가와 상담을 진행할 필요가 있다. 예를 들어, 대표적인 X-연관 열성 유전 질환인 뒤셴형 근이영양증의 경우, 남자 환자의 여자 형제(누나 또는 여동생)가 보인자인 경우가 있을 수 있는데, 보인자에서 남아를 출생할 경우에 50%의 확률로 자녀에게 동일한 질환을 가진 환자가 태어날 수 있다. 따라서 이러한 경우에는 미리 보인자 여부를 검사하여, 추후 자녀를 계획하기 이전에 해당 사실을 알려주고, 보인자로 확인되었다면 착상 전 유전 진단 또는 산전 검사 등과 같은 다양한 예방 방법을 안내해 줄 수 있다.
유전 변이의 적절한 해석으로부터 추가적인 가족 검사, 그리고 유전 상담까지 일련의 과정은 하나의 고리처럼 연결되어 있다. 유전 변이의 임상적 중요도, 해당 질환의 중증도, 사전 예방의 가능성 등을 고려하면서, 동시에 가족 관계를 생각해야 한다. 가족 검사를 통한 유전 상담은 가족 구성원에게 개인의 민감 정보를 공유하게 되므로, 가족 구성원의 개인적 상황과 윤리적, 사회적 문제 등이 관련되어 있으며, 환자와의 상호 동의가 매우 중요하다[42]. 또한 이러한 진료 과정은 소아 및 성인 희귀 유전 질환뿐 아니라, 가족성 암 질환, 그리고 임신 계획 전후의 산전 진단 등 폭넓은 임상 진료과와 연관되어 있다[43,44]. 따라서 유전자 검사 시행 이후에 대한 적절한 평가와 접근이 이루어지지 못한 채로, 마구잡이식으로 NGS 검사를 시행하는 것은 자칫 불필요한 의료 자원을 낭비함과 동시에, 환자에게 오랜 기간 진단 방랑 과정에 빠지게 하는 행위가 될 수 있음을 주지할 필요가 있다.
윤리적 측면에서의 고려 사항
의사는 환자의 유전 정보를 포함한 개인의 민감 정보를 보호해야 할 의무가 있으며, 유전 정보를 바탕으로 이를 적절하게 해석하여 올바른 진단과 치료 방침을 제공해야 한다[45,46]. 따라서 유전자 검사는 항상 환자의 자발적 요구와 동의 하에 이루어져야 하며, 유전자 검사 결과의 의미를 전달할 때는 기술적 그리고 임상적 한계를 충분히 안내하도록 하여야 한다. 특히, 환자와 가족 구성원에 대한 유전자 검사 결과가 미칠 수 있는 개인적, 사회적, 경제적 파장을 모두 신중하게 고려해야할 필요가 있다[47]. 유전자 검사 결과는 사회적 낙인으로도 작용할 수 있는데, 보험상품에 대한 가입 거절과 같은 다양한 차별 발생의 소지가 있다. 더불어 두려움을 갖고 임신 계획을 피하게 되는 등 환자와 가족에게 심리적으로도 막대한 영향을 미칠 수 있다[48]. 특히, 늦은 성인기에 발병하는 유전 질환을 대상으로 환자들의 자녀에게 유전자 검사를 시행하는 것이 필요한지에 대해서는 여전히 많은 논의가 필요하다[49]. 이 경우, 검사는 환아가 아닌 보호자의 판단에 따라 영향을 받을 수밖에 없다. 그러나 그 결과는 환아의 성인기 이후 발병 전까지 삶에 중대한 영향을 미칠 수 있어, 법적, 사회적 논의와 합의가 필요한 지점이다. 예를 들어 헌팅턴병(Huntington disease)의 경우는 40-50대 전후에 발병이 되는데, 10대 또는 20대의 이른 나이에 검사를 시행하여 유전 여부를 알게 된다면, 30여 년 동안의 시한부 선고를 받는 것과 같은 심리적 파장을 미칠 수 있다. 이로 인해 삶의 의욕을 잃거나 깊은 우울함에 시달리거나, 진로에도 영향을 줄 수 있다. 반대로, 결혼을 앞두거나 임신을 계획 중이라면 다음 자녀에게는 유전 질환을 물려주지 않도록 임신을 계획해 볼 수 있다는 점에서 도움이 될 수도 있다. 특히, 임신 중인 태아에서 진행하는 산전 유전 검사는 임신의 유지와 중단에도 영향을 미칠 수 있는 만큼, 윤리적 측면에서 더욱 신중한 고려가 필요하다[50].
최근에는 유전 질환의 진단 목적의 유전자 검사 이외에도 개인적으로 의뢰되는 다양한 유전자 검사가 점점 증가하고 있는 추세이다. 대표적으로 소비자 직접 유전자 검사(direct-to-consumer, DTC)를 통하여, 특정 유전자의 변이 또는 유전형을 검사하는 방법이 있다(Table 2) [40,41]. DTC 검사에서 대상으로 하는 유전 변이들은 인구 집단에서 빈번하게 나타나는 변이를 대상으로 하기 때문에, 유전 질환과는 관련성이 없으며 개별 변이들의 효과가 작은 경우가 대부분이다. 2023년 9월 현재까지, 최대 114개 건강 관리 항목에 대해서만 허용되고 있다[41]. 최근에는 보건복지부에서도 소비자용 가이드라인을 통하여, DTC 검사의 한계점에 대해 발표한 바 있다. 따라서 일반 소비자들은 유전자 검사법의 목적과 효용에 대해서 정확하게 인식할 필요가 있으며, 유전자 검사의 시행은 검사의 목적과 그 의미에 대한 충분한 설명과 동의 하에 시행하도록 의사들 스스로 규범적 근거를 마련할 필요가 있다.
결론
NGS 검사 방법은 다양한 유전성 질환의 진단을 위하여 효과적으로 사용할 수 있는 기술이다. 빠르게 진화하고 있는 유전자 검사 기술 속도에 발맞춰, 의사들에게도 최신의 유전적 지식 및 유전학 교육이 요구되고 있다. 유전 질환의 경우, 진단 결과가 환자의 삶에 미치는 영향이 크기 때문에, 의사들은 NGS 검사 방법의 기술적 한계를 이해하고 변이를 적절하게 판독하여 올바른 진단에 도달할 수 있어야 하며, 다학제적 접근이 도움이 될 수 있다. 가족 검사는 변이의 해석에 도움을 줄 수 있으며, 가족 구성원의 위험도 예측 및 예방, 유전 상담에도 중요한 역할을 한다. 임상 의사는 개인적, 사회적, 경제적 및 윤리적 측면을 종합적으로 판단하여, 개인 및 가족 구성원에 대한 유전자 검사를 시행하는 것이 바람직하다. 환자와 의사관계를 넘어 유전자 검사와 유전 진단이 가져올 사회적인 파장을 최소화하기 위한 공론의 장을 마련하고, 예방적 관점에서 규제 신설을 논의할 필요가 있다.