특발성폐섬유증의 진단
Diagnosis of idiopathic pulmonary fibrosis
Article information

Trans Abstract
Interstitial lung disease (ILD) is a group of diseases, involving the inflammation and fibrosis of the interstitium of the lung. ILD is classified according to whether or not the cause is known. Known causes of ILDs include inhalation of environmental substances, drugs, infection, and related connective tissue disease. ILD of unknown cause is called idiopathic ILD. The most common form of idiopathic ILD is idiopathic pulmonary fibrosis (IPF). IPF is a chronic progressive fibrosing ILD that results in the decline of lung function with exertional dyspnea, cough, bibasilar inspiratory crackles, and digital clubbing. The incidence of IPF increases with age, and is predominant in men. The most characteristic feature of IPF is a usual interstitial pneumonia (UIP) pattern detected on high-resolution computed tomography (HRCT). The typical HRCT pattern in case of UIP is honeycombing, with or without traction bronchiectasis or bronchiolectasis; this may be superimposed with fine reticulation. The typical distribution of UIP is subpleural, and there is basal predominance with heterogeneity. A definitive diagnosis of IPF in patients with clinically suspected IPF is made when there is presence of a UIP pattern on HRCT. Bronchoalveolar lavage or surgical lung biopsy is not recommended if a UIP pattern is detected on HRCT. However, bronchoalveolar lavage and surgical lung biopsy are required if probable UIP pattern, indeterminate UIP pattern, or an alternative diagnosis pattern are found on HRCT in order to diagnose IPF. A specific combination of HRCT patterns and histopathological patterns requiring multidisciplinary discussion is necessary to rule in IPF or rule it out.
서론
사이질폐질환(interstitial lung disease)은 폐사이질(lung interstitium)에 다양한 염증세포들의 침윤과 증식, 콜라겐 침착과 섬유화를 나타내는 질환들을 총칭한다[1,2]. 사이질폐질환은 원인을 알 수 없는 경우와 원인을 알 수 있는 경우로 크게 나뉜다. 원인을 알 수 없는 경우는 특발성사이질폐질환으로 분류하고, 원인을 알 수 있는 경우는 직업/환경적 요인에 의한 사이질폐질환, 의인성사이질폐질환(iatrogenic interstitial lung disease), 결체조직질환 연관 사이질폐질환으로 분류한다[1,2] (Figure 1). 직업이나 환경적 요인에 의해 발생하는 사이질폐질환으로는 분진, 화학물질, 유기물, 먼지 등의 흡인에 의해 발생하는 규폐증, 석면폐증, 베릴륨폐증, 과민성폐렴 등이 있으며, 의인성사이질폐질환은 항생제, 항암제, 항부정맥제 등의 약물이나 방사선에 의해 발생한다. 류마티스관절염, 전신홍반성낭창, 전신경화증, 쉐그렌증후군, 피부근육염, 다발근육염 등의 결체조직질환에서 발생하는 사이질폐질환은 결체조직질환 연관 사이질폐질환으로 분류한다. 그 외에도 림프관 평활근종증, 랑게르한스세포 조직구증(pulmonary Langerhans’ cell histiocytosis), 폐포단백증 등도 사이질폐질환에 속하는 폐 질환들이다[1,2].
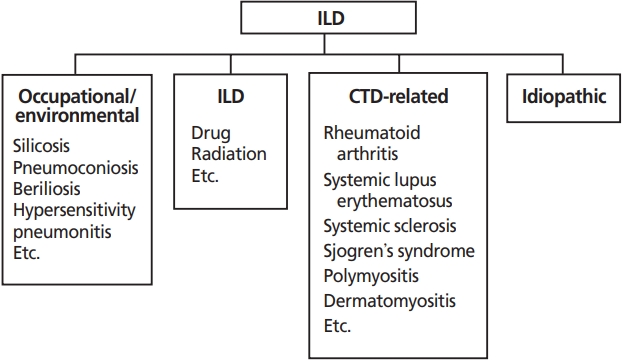
Classification of interstitial lung disease (ILD). CTD, connective tissue disease.
원인을 알 수 없는 특발성사이질폐질환의 분류는 미국 흉부학회와 유럽흉부학회의 분류기준을 사용한다[1,2] (Table 1). 주요 특발성사이질폐렴, 희귀 특발성사이질폐렴, 분류불가능 특발성사이질폐렴으로 크게 분류한다. 주요 특발성사이질폐렴에는 특발성폐섬유증, 특발성비특이성사이질폐렴, 호흡세기관지염-사이질폐질환, 박리사이질폐렴, 특발성기질화폐렴, 급성사이질폐렴이 있으며, 희귀 특발성사이질폐렴에는 특발성림프구사이질폐렴, 특발성흉막실질섬유탄력섬유증이 있다. 이 밖에 분류불가능 특발성 사이질폐렴이 있다. 이 원인 불명의 특발성사이질폐질환은 원인이 있는 사이질폐질환과 구별하기 위한 이학적 검사 및 검사실 검사, 방사선검사, 조직검사가 필요하며, 임상 의사, 영상의학의사, 병리의사들이 함께 의논하여 결정하는 다학제적 접근 진단이 필요하다[1,2]. 특발성사이질폐렴 중에서 가장 흔하면서 중증인 형태가 특발성폐섬유증(idiopathic pulmonary fibrosis)이다. 이 논문에서는 원인없이 발생하는 특발성사이질폐질환 중에서 가장 흔한 질환인 특발성폐섬유증의 진단에 대해 알아보고자 한다.
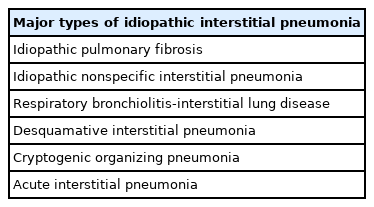
특발성폐섬유증의 진단
1. 정의
특발성폐섬유증은 원인 불명으로 발생하는 사이질 염증이 만성적으로 진행하여 폐섬유화를 보이는 질병이다. 조직 병리학적으로는 보통사이질폐렴(usual interstitial pneumonia, UIP) 소견을 보이고 있으며, 폐섬유화가 진행하므로, 점점 숨찬 증상이 심해지고, 폐기능도 감소하게 된다[3].
2. 위험인자
특발성폐섬유증은 여성보다는 남성에서 더 높은 경향을 보이고 나이가 증가할수록 증가하는 경향을 보인다. 2008년 국내 특발성사이질폐렴 전국실태조사에서도 특발성폐섬 유증은 남성이 72.4%로 많았고, 진단 시 평균나이는 69세였다[4]. 흡연을 포함한 외부환경물질의 노출이 특발성폐섬유증 발생이나 예후와의 연관성이 보고되고 있는데, 국내 실태조사에서도 특발성폐섬유증 환자의 62%가 흡연력이 있었고[4], 분진노출력이 있는 경우 특발성폐섬유증 환자들의 사망률이 증가하였다[5].
또한, 위식도 역류질환, 당뇨, 폐결핵, 비결핵 마이코박테리아폐질환(nontuberculous mycobacterial lung disease), 심장질환, 만성폐쇄성폐질환 등도 특발성폐섬유증의 발생 관련성이 제시되고 있다[6-9]. 이외에도 포진바이러스(herpesvirus) 등도 관련성이 제기되고 있으나 명확한 근거는 충분치 않다[10].
특발성폐섬유증의 위험인자로 다양한 원인들의 관련성이 제기되고는 있지만, 아직까지 그 인과관계를 증명할 근거들은 충분치 않아, 한 가지로는 원인을 설명할 수 없으며, 유전적 소인, 환경적 요인, 폐감염, 기저질환 등이 서로 복잡한 상호작용을 하여 폐간질에 염증을 유발하고 만성적으로 폐섬유화가 진행하는 것에도 기여하는 것으로 생각된다[1].
3. 임상양상
특발성폐섬유증은 원인을 알 수 없는 만성적인 진행성의 섬유화성 사이질폐렴의 형태로 고령에서 발생하고, 폐에 국한되어 발병한다. 조직학적, 방사선학적으로는 UIP 형태를 보인다. 설명되지 않는 만성적인 운동 시 호흡곤란과 기침을 호소하며, 청진음에서는 들숨 시 미세수포음이 들리고 곤봉손가락(clubbed finger)을 동반하기도 한다. 따라서 성인에서 진행하는 만성적인 운동 시 호흡곤란이나, 기침을 호소한다면 특발성폐섬유증 여부를 의심해 볼 필요가 있다.
특발성폐섬유증은 고령에서 증가하고, 대부분 6개월 이상 서서히 진행되는 호흡곤란으로 발현된다[11]. 드물지만 특발성폐섬유증 초기 발현이 급성 호흡곤란의 악화와 고해상도 컴퓨터단층촬영(high resolution computed tomography, HRCT)에서 양하엽의 기저 부위 섬유성 병변에 간유리음영(ground glass opacity, GGO)이 증가하는 급성악화로 발현되기도 한다[12]. 50대 이하에서 특발성폐섬유증의 발생은 드물기 때문에 젊은 사람의 HRCT에서 폐섬유증이 관찰된다면 결체조직질환 연관성 여부[13], 특발성폐섬유증의 가족력 여부[14], 직업에 의한 환경적 요인 등을 면밀하게 조사 및 검사해야 한다.
여성보다는 남성에 호발하고, 대부분이 흡연력이 있으므로 병력청취 시 확인이 필요하다. 또한, 위험요인으로 제시된 위식도역류질환, 바이러스감염도 확인해야 한다. 대부분의 특발성폐섬유증 환자들은 폐기종, 폐암, 폐동맥고혈압, 수면무호흡, 관상동맥질환 등의 심폐질환을 동반하고 있으므로[15] 동반 질환의 확인이 필요하다. 또한, 작업환경 등에서의 분진 노출도 위험인자가 될 수 있으므로, 직업의 종류, 종사기간, 작업환경 등에 대해서도 확인이 중요하다. 우리나라에서는 드물지만 유전적 요인도 있으므로 특발성폐섬유증 가족력 확인도 필요하다.
4. HRCT
1) 특발성폐섬유증의 HRCT 소견
특발성폐섬유증의 진단에 있어서 HRCT는 중심역할을 하는 반드시 필요한 검사이다[3]. 연속컴퓨터단층촬영 기법은 폐섬유화증의 미세하고 국소적인 폐 이상 소견까지 초기에 발견할 수 있는 장점이 있다[3]. HRCT는 총 폐용적까지 숨을 최대한 들이마신 상태로 촬영해야 한다. 숨을 최대한 들이마시지 않고 촬영하면, 폐 음영을 증가시켜 GGO나 미세 그물망 음영에 대한 평가를 잘못 할 수 있게 되므로[16], 정확한 촬영방법이 중요하다. 똑바로 누워서 HRCT를 촬영할 때 폐가 충분히 팽창되지 않고 눌려서 바닥에 가까운 폐의 음영이 증가되어 보여, 실제로는 정상임에도 불구하고 사이질폐렴의 초기처럼 오인되는 경우에는 엎드려서 찍는 prone CT가 감별에 도움이 된다[17].
특발성폐섬유증의 HRCT 소견은 전형적인 UIP 형태인 벌집모양(honeycombing) 변화, 견인성 기관지확장증 또는 견인성 세기관지확장증과 미세한 그물망 음영 변화를 보이며, 간혹 GGO가 동반되기도 한다(Figure 2) [3]. 이런 특발성폐섬유증 병변의 영상학적 소견은 폐 양쪽의 기저부, 늑막하, 말초에서 시작되어 위치한다[18].

High-resolution computed tomography (CT) images of a usual interstitial pneumonia pattern. Transverse CT section (A) and coronal CT section (B) illustrate subpleural and basal predominant honeycombing comprising clustered cycts with well-defined walls and variable sizes. Fine reticulation (red arrow) and bronchiectasis (blue arrow) were also observed. Informed consent for publication of the clinical image was provided by the patient.
벌집모양의 변화는 폐실질이 3-10 mm의 낭포성 공간으로 바뀌는 것으로, 특발성폐섬유증 진단에 중요한 단서이다. 이 낭포는 더 커질 수도 있으며, 낭포벽이 두꺼워 구별이 잘 되는 특징을 보인다[19]. 대부분 여러 층의 낭포가 관찰되어 벌집모양처럼 모여 벌집모양 변화라고 하지만, 여러 층이 아닌 단일층으로 관찰되는 경우도 있는데 이럴 때는 폐기종과 구별할 필요가 있다. 이 벌집모양 변화는 그물망 변화와 견인성 기관지확장증 또는 세기관지확장증을 동반한다[20].
견인성 기관지확장증과 세기관지확장증은 폐섬유화의 특징이다. 폐실질이 섬유화 되면서 오그라들어 주변의 기관지가 따라오면서 늘어나서 발생한다[19]. 특발성폐섬유증의 견인성 기관지확장증은 주로 폐 말단 부위와 늑막하 부위에 위치하며 그 주변은 염증에 의한 그물망 음영 병변과 GGO가 같이 관찰된다[21].
그물망 음영 소견은 미세한 선들의 망이다. UIP에서 그물망 음영 소견은 불규칙한 굵은 선과 얇은 선의 망으로 관찰된다[19]. GGO는 기관지와 혈관의 형태는 잘 유지되면서 폐음영이 뿌옇게 증가된 것이다[19]. 특발성폐섬유증의 GGO는 미세 그물망 음영 소견과 같이 관찰되며, 견인성기관지확장증 주변에서 관찰된다[3]. 미세 그물망 음영 소견 없이 GGO만 관찰되는 경우는 UIP의 전형적인 소견은 아니다. 만일 특발성폐섬유증 환자에서 미세 그물망 음영 소견 없이 GGO만 관찰된다면 특발성폐섬유증의 급성악화를 시사한다[22].
2) HRCT 형태에 따른 분류
특발성폐섬유증을 진단하기 위해 HRCT 형태는 다음의 네 가지로 분류한다[3] (Table 2). 첫째, UIP 형태이다. UIP는 특발성폐섬유증의 방사선학적 형태의 특징이다. 벌집모양은 UIP를 구별하는 특징적인 소견으로 HRCT의 UIP 진단에 필수적이다. 벌집모양은 말초 견인성 기관지확장증이나 세기관지확장증을 동반하기도 한다. 병변은 기저부 늑막하 부위에서 주로 나타나는 게 특징이나, 상엽에 나타나기도 한다. CT에서 UIP소견인 경우, 조직에서도 UIP 소견을 보일 양성 예측률은 90-100%이지만, 일부에서는 조직병리학적으로 UIP이지만 HRCT는 UIP 형태가 아닌 경우도 있다[3].
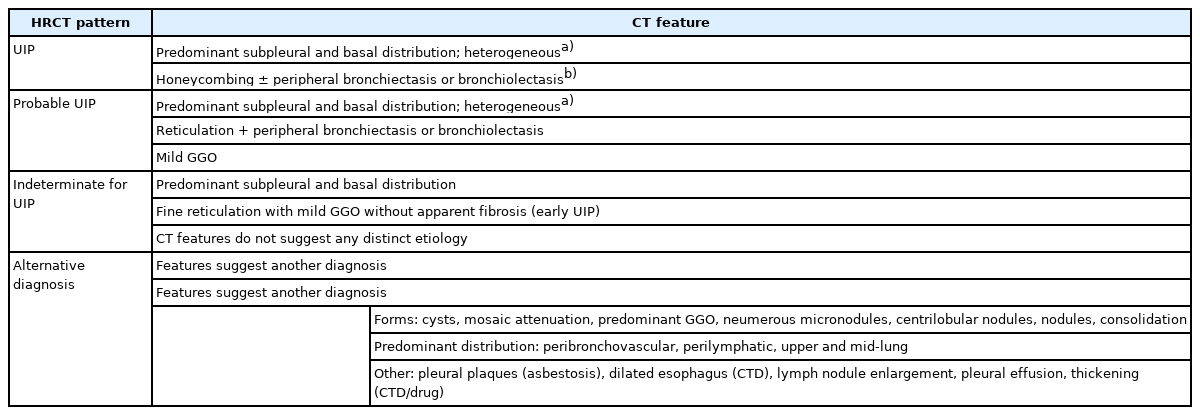
Classification of HRCT patterns and features according to the official ATS/ERS/JRS/ALAT clinical practice guideline
둘째, probable UIP 형태이다. 과거 possible UIP의 CT는 벌집모양 없이 기저부 늑막하 부위 그물망 음영의 형태로 정의했으나[23], 이 형태는 조직소견에서 UIP와 높은 일치율을 보임에 따라[24], 말단부 견인성 기관지확장증 또는 세기관지확장증을 동반하는 기저부 위주의 그물망 음영 형태를 “probable UIP” 로 재정의 하였다. GGO는 동반할 수 있으나 주된 소견은 아니다[3].
셋째, indeterminate for UIP 형태이다. 과거 비전형적인 HRCT 소견을 보인 경우의 약 30%는 조직에서 UIP 형태를 보였다[25]. 따라서, indeterminate 형태는 HRCT에서 UIP나 probable UIP를 만족하지 못하면서, 명확하게 다른 진단 소견이 없이 섬유화 소견이 있는 경우로 정의한다. 늑막하 부위에 국한된 GGO와 명백한 섬유화 없이 그물망 음영 소견이 관찰되기도 한다. 초기 UIP나 probable UIP에서 관찰되기도 하는데, 이럴 경우 prone CT를 촬영하면 의존성 폐허탈에 의한 GGO와 구별할 수 있다[3].
넷째, alternative diagnosis이다. Table 2에 alternative diagnosis에 소개된 CT 소견들은 특발성폐섬유증이 아닌 다른 진단을 고려해야 한다[3].
5. 폐 조직검사
HRCT에서 UIP의 특징적인 소견을 보이면서 특별한 원인이 없다면, 특발성폐섬유증 진단을 위한 폐 조직검사는 필요하지 않다. HRCT에서 probable UIP, indeterminate for UIP, alternative diagnosis인 경우엔 수술적 폐생검을 시행해서(Figure 3), 조직병리학적 형태에 따라(Table 3) HRCT 형태(Table 2)와 특정조합하여 진단하도록 한다(Table 4) [3]. 수술적 폐생검 시 한쪽 폐 환기가 가능한 경우라면 개흉 폐 생검보다는 흉강경을 이용한 폐 생검을 선호한다. 폐기능저하가 심하고 동반질환이 심하다면 수술적 폐생검은 이득보다는 해가 더 많을 수 있으므로 환자의 상태에 따라 결정해야 한다. 수술적 폐생검은 서로 다른 엽에서 여러 개 시행한다[3]. 특발성폐섬유증 진단에 폐생검이 필요할 경우 냉동 폐 생검이나 기관지경유 폐생검으로의 대치에 대해서는 아직 합의된 권고사항은 없다[3].
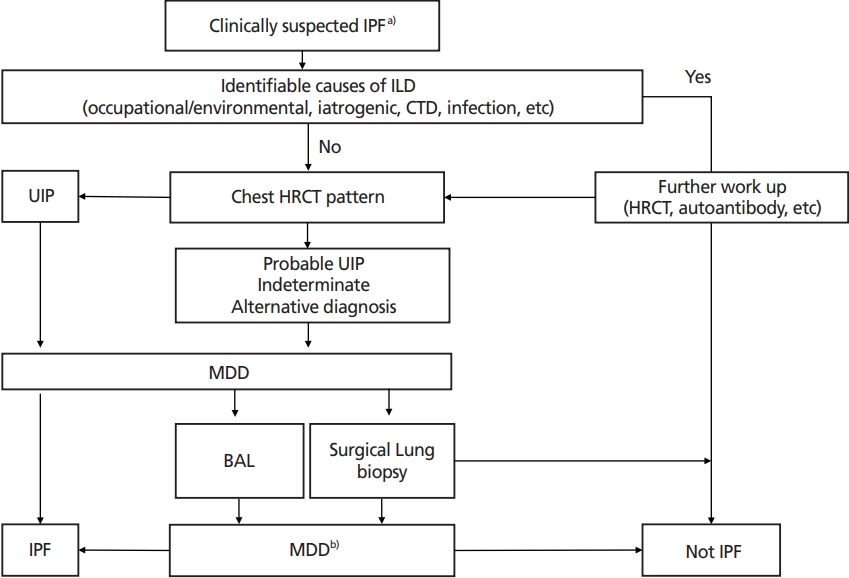
Diagnostic algorithm for idiopathic pulmonary fibrosis (IPF). ILD, interstitial lung disease; CTD, connective tissue disease; HRCT, high-resolution computed tomography; UIP, usual interstitial pneumonia; MDD, multidisciplinary discussion; BAL, bronchoalveolar lavage. a)Clinically suspected IPF: patients with unexplained symptomatic or asymptomatic bilateral pulmonary fibrosis on chest X-ray or HRCT scan, bibasilar inspiratory crackles, and age typically older than 60 years (40–60 years in case of a family history of pulmonary fibrosis) [3]. b)Specific combination of HRCT patterns and histopathology patterns during MDD for ascertainment or exclusion of IPF diagnosis [3].

Specific combinations of HRCT patterns and histopathologic patterns for diagnosis of IPF
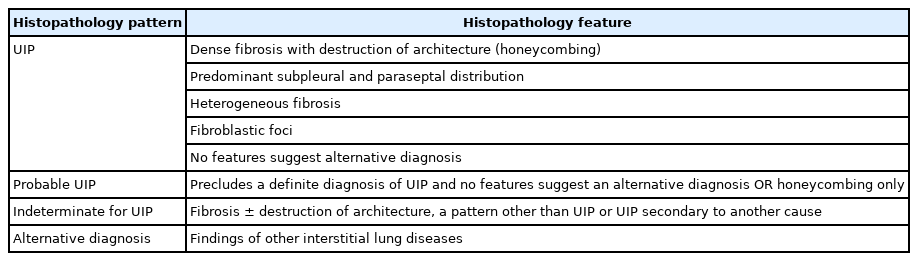
6. UIP의 조직병리학적 특징
UIP의 조직병리학적 특징은 저배율 소견에서 조밀한 섬유화가 부분적으로 관찰되는데, 전형적으로 늑막하 격벽 주변 폐실질을 침범한다. 이 섬유화로 인해 폐 구조는 리모델링되고, 벌집모양이 생기게 된다. 또한, 사이질 림프구와 형질세포의 침윤에 의한 사이질 염증이 관찰되고 2형 폐포세포와 기관지 상피세포의 과증식을 동반한다. 섬유화 병변에서는 조밀한 콜라젠의 침착과 섬유모세포와 근섬유모세포의 증식병변인 섬유모세포병소가 관찰되는게 특징이다. 현미경 상의 벌집모양은 폐포가 섬유화성 낭종으로 변화된 것인데 이는 세기관지 상피세포로 덮이고 가래와 염증세포로 채워지며, 평활근 이형성이 간질에서 관찰된다. 이런 섬유화 병변과 벌집모양, 염증병변은 정상소견과 혼재되어 동시에 관찰되는 이질성 특징을 보인다. 이런 모든 병변이 관찰되면 UIP 형태로 진단하고, 특히 벌집모양이 있으면 특징적이지만, 벌집 모양이 없이 다른 소견들만 있어도 UIP 진단은 가능하다. 이런 조직소견들은 과민성폐렴, 특발성기질화폐렴, 진폐증, 유육종증 등 다양한 다른 진단들을 배제하는데 도움이 된다. 또한, HRCT 소견이나 조직병리학적 소견에서 UIP소견은 특발성폐섬유증에서만 보이는 소견은 아니다. 결체조직질환 관련 UIP도 있으므로 동반질환의 유무, 약제 복용력, 환경노출력, 가족력 등을 세밀하게 확인하는 것이 중요하다.
UIP 조직병리학적 소견도 HRCT 형태처럼 UIP, probable UIP, indeterminate UIP, alternative diagnosis 네 개의 형태로 구별한다(Table 3) [3]. HRCT와 조직병리학적 형태를 같은 용어로 분류한 것은 다학제적 접근 시 토론을 수월하게하며, 적절한 진단 도출에도 도움이 된다[3].
7. 특발성폐섬유증 진단기준
특발성폐섬유증의 진단은 임상소견, 흉부방사선학적 소견, 병리조직학적 소견을 종합하여 고려해야 한다. 이를 위해서는 호흡기내과, 영상의학과, 병리과 의사의 다학제 진료가 필요하다. 2018년 미국, 유럽, 일본, 라틴아메리카 흉부학회들이 공동으로 발표한 특발성폐섬유증 진단 임상지침서에 따르면, 특발성폐섬유증 진단을 위해서는 1) 다른 원인에 의한 사이질폐질환(직업환경적 노출, 결체조직질환, 약물 등에 의한 사이질폐질환)을 배제해야 하고, 2) HRCT에서 UIP 형태가 있어야 하며(Table 2), 3) 폐생검 조직검사를 시행한 환자라면 HRCT 형태(Table 2)와 조직병리학적 형태(Table 3)의 특정조합(Table 4)을 이용할 것을 제시하고 있다[3].
증상의 유무와 상관없이 양측의 폐섬유화 소견이 가슴 X선이나 CT에서 관찰되고, 양쪽 폐 기저부에서 들숨 수포음이 청진 되며, 60세 이상인 경우와 같이 임상적으로 특발성폐섬유증이 의심될 때, 가장 먼저 환경적인 노출력이나 직업력, 결체조직질환, 약물, 방사선 등 사이질폐질환의 다른 원인이 있는지 확인한다. 단, 특발성폐섬유증의 가족력이 있는 경우에는 40-60세에도 발현하므로 60세 이상으로 제한을 두지 않는다. 만일 원인이 있는 사이질폐질환이 의심된다면 해당 원인질환 및 관련된 사이질폐질환의 진단과 배제를 위한 검사를 한다. 사이질폐질환을 유발할 만한 원인이 없다면 HRCT 형태에 대해 다학제 진료를 통해 특발성폐섬유증 여부를 의논한다(Figure 3). 만일 젊은 50세 이하, 특히 여성에서 특발성폐섬유증이 진단되었다면, 진단 이후에 결체조직질환의 임상적증상이나 혈청학적 소견이 나타나는 경우도 있으므로, 젊은 연령에서는 세심한 관찰과 추적이 필요하다[1].
사이질폐질환을 유발할 특정 원인이 없고, HRCT에서 UIP 형태가 보인다면 특발성폐섬유증으로 진단하며, 특발성폐섬유증 확진을 위한 추가적인 기관지폐포세척술이나 수술적 폐생검은 필요하지 않다[1]. HRCT에서 probable UIP, indeterminate for UIP, alternative diagnosis 소견이라면 다학제 진료, 기관지폐포세척술, 수술적 폐생검을 통해 특발성폐섬유증을 진단한다(Figure 3). 이때 HRCT와 조직병리학적 형태를 특정조합하여 이용한다(Table 4) [3]. 폐생검이 필요할 경우 환자의 상태를 고려하여 결정한다[3].
결론
원인 미상의 특발성폐섬유증은 특발성사이질폐렴 중에서 가장 흔하다. 남성, 60세 이상에서 호발하고, 흡연 및 분진 노출, 위식도역류질환, 당뇨, 심장질환, 만성폐쇄성폐질환, 바이러스감염, 유전적소인 등이 위험인자로 제시되고 있으며, 50세 이하에서도 가족력과 관련하여 발생할 수 있다. 따라서 원인 유무를 구별하기 위한 병력청취가 중요하다.
임상양상은 원인 불명의 6개월 이상의 만성 호흡곤란, 기침, 들숨 수포음이 특징이다. 특발성폐섬유증의 전형적인 HRCT 특징은 양쪽 폐 기저부 늑막하 부위에 병변이 주로 위치하며, 이 병변은 UIP 소견인 벌집모양의 구조와 기관지확장증·세기관지확장증을 보이고, 미세한 그물망 음영과 경미한 GGO를 동반할 수 있다. 이런 이질성 분포의 UIP의 특징적인 HRCT 형태를 보이면 추가적인 기관지폐포세척술과 폐생검 없이 특발성폐섬유증을 진단한다. 특징적인 HRCT 형태를 보이지 않는다면, 폐생검을 시행해서 HRCT와 조직병리학적 특징을 특정조합을 이용해 특발성폐섬유증 여부를 진단한다. 이때 다학제적 접근이 정확한 진단에 도움이 된다.
HRCT 소견과 조직병리학적 소견에서 UIP라고 하더라도 결체조직연관 UIP일 수도 있으므로, 결체조직연관 관련여부를 확인하기 위해 병력청취 뿐만 아니라, 신체검사와 자가항체검사도 중요하다. 특발성사이질폐질환은 그 진단에 따라 치료와 예후가 다르므로 정확한 진단을 위한 노력이 중요하다.
Notes
Conflict of Interest
No potential conflict of interest relevant to this article was reported.
References
Peer Reviewers’ Commentary
사이질폐질환은 폐 사이질의 증식, 염증세포 침윤, 때로는 섬유화를 동반하는 여러 폐질환들을 포괄한다. 사이질폐질환의 임상적, 영상의학적, 병리학적 특성이 매우 다양하기 때문에 임상 진단이 쉽지 않다. 이 논문에서는 사이질폐질환의 분류 개념과 함께 대표적인 질환인 특발성폐섬유증의 진단 기준을 2018년 개정된 국제진단지침 및 대한결핵및호흡기학회 사이질폐질환 임상진료지침을 토대로 소개하고 있다. 특발성폐섬유증의 HRCT 소견과 병리소견, 어떤 경우에 진단을 위해 수술적 폐생검이 필요한 지에 대해 자세히 설명하고 있다. 진단지침에 따라 원인을 찾고, HRCT소견을 고려하여 다학제 진료 후 수술적 폐생검을 결정하고, 다시 최종적으로 호흡기내과, 영상의학과, 병리과 의사가 다학제 진료를 통해 진단에 이르는 과정을 잘 설명하고 있다. 이 논문은 임상에서 특발성폐섬유증을 진단하는데 좋은 지침을 제공해 줄 것으로 판단된다.
[정리: 편집위원회]